Теория резонанса
Концепция (или теория) резонанса была предложена Полингом в начале 30-х годов. Основная идея ее заключалась в следующем. Если Ψ0 представляет некоторую волновую функцию системы, то интеграл 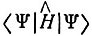 (Н^ - оператор Гамильтона) должен быть больше или равен энергии наинизшего состояния Е0. Чем ближе Ψ к этой собственной функции, тем меньше будет разность Е - Е0. Допустим, что мы нашли функцию Ψ1, которая представляет возможные состояния системы, например состояние, соответствующее некоторой электронной формуле Льюиса. Тогда при замене Ψ на Ψ1 в указанном интеграле можно рассчитать электронную энергию E1, как функцию от межъядерных расстояний. Аналогично функция Ψ2, соответствующая альтернативной электронной формуле, может быть использована для расчета Е2. Если уровень Е1 лежит значительно ниже уровня E2, то функция Ψ1 будет лучше аппроксимировать основное состояние системы, чем Ψ2, и если другие альтернативы отсутствуют, то можно принимать во внимание только электронную формулу, соответствующую Ψ1. Вообще говоря, если Ψ1 и Ψ2 имеют одинаковый характер симметрии и, что особенно важно, одинаковую мультиплетность (т. е. одно и то же число неспаренных электронов), то может быть найдено значение E, соответствующее функции aΨ1 + bΨ2. Когда Е1 и E2 не очень различаются и когда члены, соответствующие взаимодействию между состояниями Ψ1 и &Ψ2 велики, то оказывается, что функцией, дающей наилучшее приближение к собственной функции основного состояния системы, будет не Ψ1 и не Ψ2, а их линейная комбинация с коэффициентами а и b, являющимися величинами одного порядка. В этом случае ни одна электронная формула сама по себе не может быть сопоставлена молекуле. Необходимы обе структуры, хотя одна из них, возможно, будет иметь больший вес, чем другая. "Молекула,- отмечает Полинг,- могла бы рассматриваться как быстро флуктуирующая между двумя электронными формулами, и ее стабильность получается большей, чем для любой из этих формул благодаря "энергии резонанса этих флуктуации" [72, с. 996-997]. Впоследствии теория резонанса была развита Полингом, Уэландом и другими авторами, которые применили ее к широкому кругу химических соединений.
(Н^ - оператор Гамильтона) должен быть больше или равен энергии наинизшего состояния Е0. Чем ближе Ψ к этой собственной функции, тем меньше будет разность Е - Е0. Допустим, что мы нашли функцию Ψ1, которая представляет возможные состояния системы, например состояние, соответствующее некоторой электронной формуле Льюиса. Тогда при замене Ψ на Ψ1 в указанном интеграле можно рассчитать электронную энергию E1, как функцию от межъядерных расстояний. Аналогично функция Ψ2, соответствующая альтернативной электронной формуле, может быть использована для расчета Е2. Если уровень Е1 лежит значительно ниже уровня E2, то функция Ψ1 будет лучше аппроксимировать основное состояние системы, чем Ψ2, и если другие альтернативы отсутствуют, то можно принимать во внимание только электронную формулу, соответствующую Ψ1. Вообще говоря, если Ψ1 и Ψ2 имеют одинаковый характер симметрии и, что особенно важно, одинаковую мультиплетность (т. е. одно и то же число неспаренных электронов), то может быть найдено значение E, соответствующее функции aΨ1 + bΨ2. Когда Е1 и E2 не очень различаются и когда члены, соответствующие взаимодействию между состояниями Ψ1 и &Ψ2 велики, то оказывается, что функцией, дающей наилучшее приближение к собственной функции основного состояния системы, будет не Ψ1 и не Ψ2, а их линейная комбинация с коэффициентами а и b, являющимися величинами одного порядка. В этом случае ни одна электронная формула сама по себе не может быть сопоставлена молекуле. Необходимы обе структуры, хотя одна из них, возможно, будет иметь больший вес, чем другая. "Молекула,- отмечает Полинг,- могла бы рассматриваться как быстро флуктуирующая между двумя электронными формулами, и ее стабильность получается большей, чем для любой из этих формул благодаря "энергии резонанса этих флуктуации" [72, с. 996-997]. Впоследствии теория резонанса была развита Полингом, Уэландом и другими авторами, которые применили ее к широкому кругу химических соединений.
Достаточно подробно генезис этой теории был рассмотрен в монографии Г. В. Быкова [5]. Поэтому, избегая повторений, мы остановимся в дальнейшем только на тех вопросах, которые не были должным образом освещены в литературе.
Наибольшее распространение концепция резонанса нашла в органической химии. При этом популярность ее была так велика, что она часто отождествлялась с методом ВС. Когда же гипертрофирование роли резонанса электронных структур было подвергнуто критике, такое отождествление отрицательно сказалось на отношении многих химиков к методу ВС и привело к неправильному пониманию роли и логической структуры последнего. Историческое значение концепции резонанса состоит, во-первых, в том, что она определила одно из возможных направлений развития метода ВС. Во-вторых, она позволила глубже понять соотношение между классической и квантовой теориями строения химических соединений, вскрыв те стороны физической и химической реальности, которые не могли быть адекватно отражены классической теорией строения.
Чтобы яснее представить роль резонанса в логической структуре этого метода, попытаемся ответить на следующий вопрос: возможен ли "безрезонансный" метод ВС, и если да, то каковы будут его особенности. С ретроспективной точки зрения иной возможный путь развития метода ВС мог состоять в сохранении приближения идеального спаривания, но при этом пришлось бы обобщить концепцию гибридизации, т. е. использовать в качестве базисных функций не атомные, пусть даже гибридные (в обычном смысле слова) орбитали, а их линейные комбинации, вообще говоря, не ортогональные*. Уравнениями, определяющими эти линейные комбинации, являются уравнения Годдарда [43]. В некотором смысле этот метод, названный Годдардом "обобщенным методом ВС", является одновременно и обобщением метода МО. Иными словами, концепция резонанса служила не только одним из способов выражения метода ВС, который придал мышлению химиков большую гибкость, но и явилась своеобразным водоразделом, отделяющим два наиболее распространенных метода квантовой химии, ВС и МО, так как "безрезонансный" вариант метода ВС представляет собой такую модификацию последнего, которая придает ему черты метода МО.
* (Базисные АО в обычной формулировке метода ВС также не ортогональны.)
Проиллюстрируем этот тезис на примере молекулы бензола. В методе ВС для описания π-электронной системы молекулы бензола необходим учет пяти независимых структур, характеризуемых диаграммами I-V (см. рис. 16). Эти диаграммы могут быть построены с использованием схем и таблиц Юнга.
Антисимметричная собственная функция оператора 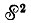 может быть получена из произведения координатной Φ и спиновой Χ функций действием оператора Годдарда
может быть получена из произведения координатной Φ и спиновой Χ функций действием оператора Годдарда 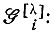
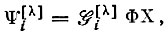 (3.50)
(3.50)где
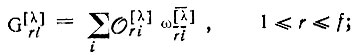 (3.51a)
(3.51a)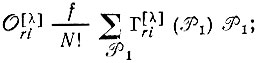 (3.51б)
(3.51б)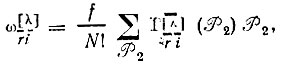 (3.51в)
(3.51в)
где 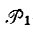 - операторы перестановки пространственных координат;
- операторы перестановки пространственных координат; 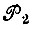 - операторы перестановки спиновых переменных;
- операторы перестановки спиновых переменных; 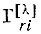 - матричные элементы неприводимого представления [λ] группы перестановок N-электронов; f - размерность этого представления.
- матричные элементы неприводимого представления [λ] группы перестановок N-электронов; f - размерность этого представления.
В методе Годдарда используется специальный выбор функций Φ и Χ в виде произведений одноэлектронных функций:
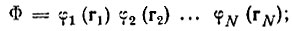 (3.52)
(3.52)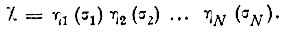 (3.53)
(3.53)
Многоэлектронной волновой функции метода Годдарда можно сопоставить определенную схему спинового спаривания, которой будет соответствовать некоторая обобщенная диаграмма Румера*. Действительно, как показал Годдард, действие оператора 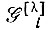 на произведение Φ и Χ эквивалентно действию оператора Юнга
на произведение Φ и Χ эквивалентно действию оператора Юнга 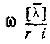 на X с последующей антисимметризацией:
на X с последующей антисимметризацией:
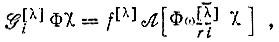 (3.54)
(3.54)что при соответствующем выборе X полностью соответствует построению многоэлектронной функции метода ВС. Например, для π-электронной системы бензола выбору
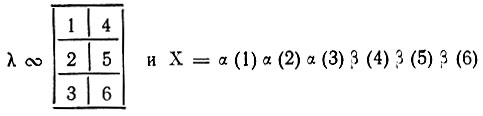
будет соответствовать схема спинового спаривания, выражаемая следующей диаграммой:
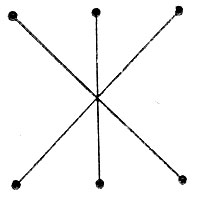 (3.56)
(3.56)* (Под обобщенной мы понимаем такую диаграмму Румера, которая Может содержать перекрещивающиеся штрихи.)
Таким образом, вместо пяти диаграмм в обобщенном методе ВС мы имеем только одну. Эта диаграмма совпадает по внешнему виду с диаграммой V (см. рис. 16). Однако в то время как диаграммы I-V характеризуют спаривание атомных π-орбиталей, в диаграмме (3.56) спаренными следует считать линейные комбинации последних (молекулярные орбитали) φk, которые определяются уравнениями вида*
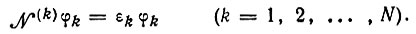 (3.57)
(3.57)* (В методе МО молекулярные орбитали удовлетворяют аналогичным уравнениям, но с общим для всех k эффективным гамильтонианом N, что обусловливает их ортогональность. В методе Годдарда орбитали не ортогональны и в этом отношении напоминают атомные орбитали.)
Существенно, что этим уравнениям может быть дана интерпретация в рамках модели независимых частиц (МНЧ), т. е. отдельному электрону можно приписать определенное состояние, характеризуемое орбиталью φk. Следуя Годдарду, можно указать три условия, обеспечивающие возможность такой интерпретации:
- N электронам сопоставляется не более чем N различных орбиталей;
- каждая орбиталь должна быть собственной функцией некоторого эффективного гамильтониана, определяющего движение электрона в поле ядер и в усредненном поле других электронов;
- это усредненное поле может быть нелокальным, но оно должно быть самосогласованным.
В отличие от метода Годдарда метод ВС в своей обычной формулировке не удовлетворяет условиям (2) и (3) и поэтому не может быть интерпретирован в терминах МНЧ. В то же время он допускает обобщение в рамках метода Годдарда, удовлетворяющее всем трем указанным выше условиям, в силу чего его интерпретация в терминах МНЧ становится возможной.
Разумеется, в начале30-х годов (и позже) сформулированный выше подход не мог быть реализован, главным образом, потому, что ввиду отсутствия необходимой вычислительной техники теория развивалась в основном на базе полуэмпирических и эмпирических методов, а также интуитивного обобщения методов, развитых для простых систем и близких (по крайней мере, в семантическом плане) классической теории строения. Конечно, отсутствие вычислительной техники, обеспечивающей преодоление математических трудностей многоэлектронной задачи, при стремлении к прогрессу в понимании электронной структуры атомов и молекул способствовало развитию фундаментальных концепций, сохранивших свое значение и до настоящего времени. Однако при этом наибольшее развитие получали те идеи и методы, которые могли плодотворно использоваться в условиях существования большого разрыва между качественными и количественными сторонами теории.
Обратимся теперь к другому вопросу - о реальности резонансных структур. Сначала несколько замечаний о терминологии. Мы считаем, что термин "резонансные структуры" можно применять лишь в том случае, если речь идет об эквивалентных структурах метода ВС. Например, нельзя называть резонансными структуры бутадиена 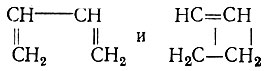 или циклооктатетраена
или циклооктатетраена 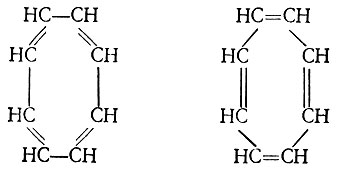 .
.
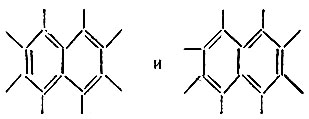
В каждом из этих примеров первая структура может использоваться в качестве структурной формулы соединения, а вторая - не может, так как ее вес пренебрежимо мал. Действительно, длина одинарной связи в такой структуре оказывается меньше, чем длина двойной, что противоречит известным эмпирическим закономерностям, связывающим кратность связи с ее длиной. О резонансе и резонансных структурах имеет смысл говорить, когда соответствующие этим структурам квантовомеханические средние значения энергии* равны или близки. Однако не следует связывать резонанс, понимаемый в указанном выше смысле, с какими-либо колебаниями, осцилляциями, пульсациями или флуктуациями, как это делал Полинг и другие авторы. Такие псевдоклассические представления, имеющие сомнительную ценность в отношении электронной системы молекулы, совершенно ошибочны в отношении атомных ядер, которые на данном уровне рассмотрения (электронная задача в адиабатическом приближении) следует считать неподвижными. В случае "резонанса" структур соединение обычно нельзя охарактеризовать классической структурной формулой, которая не противоречила бы его свойствам. Например, для бензола ни одна из двух классических формул Кекуле не отражает симметрии молекулы, ее физических и химических свойств. Аналогично формула 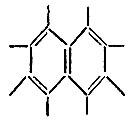 не является вполне адекватной для молекулы нафталина, так как следует принимать во внимание еще хотя бы две структуры:
не является вполне адекватной для молекулы нафталина, так как следует принимать во внимание еще хотя бы две структуры:
* (Эти значения электронной энергии определяются при фиксированной и тождественной для всех структур конфигурации атомных ядер методами квантовой химии. Они не имеют непосредственного физического или химического смысла и не измеряются экспериментально.)
Резонанс структур в органической химии обычно обусловлен сопряжением одинарных и двойных связей углерод-углерод, особенно в плоских циклических системах (ароматические углеводороды и гетероциклы). Поэтому концепция резонанса некоторое время лежала в основе теории таких соединений, пока ее не сменил метод МО ЛКАО.
Иногда с понятием о резонансных структурах связывают понятие об "электронных изомерах". При этом их определяют как химические соединения, характеризуемые одной и той же ядерной конфигурацией, но различным распределением электронной плотности. Такое представление является безусловно ошибочным, так как именно распределение электронной плотности и определяет равновесную ядерную конфигурацию. Электронным изомерам поэтому неизбежно должны соответствовать различные ядерные конфигурации, так что это понятие сводится к обычному понятию изомерии (см. подробнее работу [2]).
В свете сказанного выше закономерен вопрос: какие же стороны объективной реальности отражает концепция резонанса?
Необходимость учета нескольких резонансных структур связана прежде всего с тем, что не всегда возможно приписать химическую связь отдельным парам атомов, т. е. химическая связь может быть делокализована между тремя и большим числом атомов. Такой делокализации соответствует резонанс ковалентных структур. В то же время в соединениях с локализованными двухцентровыми связями последние могут быть (и обычно являются) поляризованными. Для отражения полярности связи следует учитывать ионно-ковалентный резонанс. В некоторых случаях без учета резонанса структур может получаться качественно неправильное описание электронной структуры молекулы, в частности, может нарушаться соответствие между симметрией молекулы и распределением в ней электронной плотности, примером чему может служить молекула бензола. Одноструктурное представление соединения, принятое в классической теории химического строения, является приближенным с точки зрения квантовохимической теории, описывающей строение химических соединений (в рамках метода ВС) несколькими резонансными структурами. Иными словами, понятие резонанса на уровне приближения, определяемом методом ВС, в концентрированном, предельно схематичном виде отражает всю эволюцию теории химического строения - от приписывания каждому индивидуальному соединению определенной классической структурной формулы до учета делокализации электронов в квантовой теории. Тем самым в принципиальном отношении появление концепции резонанса исторически явилось завершением круга идей, лежащих в основе метода ВС.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'