Квантовомеханичеекая интерпретация молекулярных спектров и ее роль в создании метода молекулярных орбиталей
Основные положения квантовомеханической теории молекулярных спектров были сформулированы в серии статей Хунда, опубликованных в 1927-1930 гг. под общим заголовком "К интерпретации молекулярных спектров" [54]. В них были заложены основы метода молекулярных орбиталей - доминирующего метода расчета электронной структуры молекул в современной квантовой химии. В первой из статей указанной серии - [54, I], преследуя цель качественного объяснения природы молекулярных спектров, Хунд рассмотрел простейшую модель молекулы - квантовомеханическую систему с одной степенью свободы, потенциальная энергия которой характеризуется существованием нескольких минимумов. Существенным является то, что при этом он отметил возможность установить соответствие между стационарными состояниями рассматриваемой модельной системы и состояниями, которые отвечают бесконечному удалению потенциальных минимумов друг от друга. Тем самым была установлена адиабатическая взаимосвязь между состояниями двух разделенных атомов или ионов, состояниями двухатомной молекулы и состояниями атома, образованного путем мысленного сближения атомов вплоть до объединения их ядер. Ранее аналогичные идеи высказывали некоторые авторы (например, Кондон), но они были сформулированы недостаточно четко. Практическая ценность отмеченных Хундом корреляций состояла в том, что они позволили во многих случаях получить качественно правильную схему взаимного расположения энергетических термов двухатомной молекулы.
Работа Хунда [54, II] была посвящена исследованию характерных свойств полосатых спектров двухатомных молекул (как гомо-, так и гетеронуклеарных). В дальнейшем наряду с электронной он рассматривал также колебательную и вращательную структуры этих спектров [54, III-V]. Мы остановимся только на первых двух работах, наиболее повлиявших на процесс создания молекулярно-орбитальной теории молекул.
Согласно Хунду, электронную систему двухатомной молекулы можно представить как построенную путем последовательного добавления в поле двух атомных ядер по два электрона. При этом возникает вопрос: какое квантовое состояние займет каждый из добавляемых электронов, т. е. какова последовательность одноэлектронных квантовых состояний? Очевидно, что она зависит как от зарядов атомных ядер, так и от расстояния между ними. Хунд рассматривает два случая - малые и большие межъядерные расстояния R.
Если R мало по сравнению с эффективными размерами электронных оболочек атомов, то молекулярные термы должны быть подобны термам атомным*. При этом атомному Р-терму будут соответствовать два близких по энергии 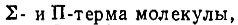 атомному D-терму - три молекулярных:
атомному D-терму - три молекулярных: 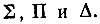
* (Связь молекулярных термов с атомными была рассмотрена в 1928 г. также Вигнером, но в более общем виде с помощью теории групп.)
Одноэлектронные состояния образуют при малых R ту же последовательность, что и в атоме: 1s, 2s, 2p, 3s, 3р, 4s, 3d,..., если система электронейтральна или ее заряд мал; и 1s, 2s, 2p, 3s, 3р, 3d, 4s,..., если суммарный заряд ядер существенно больше числа электронов.
Простейшим случаем, рассмотренным Хундом, является атом, содержащий замкнутые электронные оболочки и один р-электрон в незамкнутой оболочке. Такой атом находится в состоянии 2Р. Мысленное расщепление ядра приводит к понижению сферической симметрии до аксиальной и, следовательно, к расщеплению 2Р-терма на 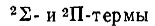 при допущении, что 2∏-терм лежит выше терма 2∑.
при допущении, что 2∏-терм лежит выше терма 2∑.
При наличии сверхзамкнутой оболочки лишь одного d-электрона 2D-терм объединенного атома порождает молекулярные 2∏-, 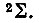 - и 2Δ-термы, приведенные здесь в порядке возрастания их энергии. При наличии пяти эквивалентных р-электронов соответствующий 2Р-терм порождает
- и 2Δ-термы, приведенные здесь в порядке возрастания их энергии. При наличии пяти эквивалентных р-электронов соответствующий 2Р-терм порождает 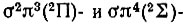 -состояния, причем последнее имеет большую энергию. При добавлении еще одного электрона из двух указанных выше термов, 2∏ и 2∑, возникает терм
-состояния, причем последнее имеет большую энергию. При добавлении еще одного электрона из двух указанных выше термов, 2∏ и 2∑, возникает терм 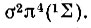
Приведенные выше рассуждения Хунда относились к случаю, когда расстояние между ядрами являлось достаточно малым, чтобы расщепление атомных термов было существенно меньше, чем расстояние между ними на шкале энергии. Если теперь несколько увеличить межъядерное расстояние и (одновременно) взаимодействие электронов считать несколько меньшим, то энергетическая последовательность электронных уровней будет определяться в первую очередь квантовыми числами n и l, во вторую очередь - квантовым числом |m| и только в третью очередь - квантовыми числами полного спина и абсолютной величиной проекции полного орбитального момента импульса на ось молекулы. Последовательность одноэлектронных состояний характеризуется тогда рядом
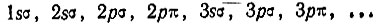
Обратимся теперь к рассмотренному Хундом случаю разделенных атомов. При достаточном разведении атомных ядер термы двухатомной молекулы должны перейти в атомные термы. Если заряды ядер одинаковы (гомонуклеарная молекула), то атомные орбитали могут порождать молекулярные орбитали согласно схеме:
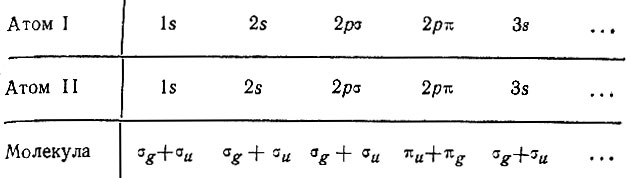
Атомные 1s-уровни при сближении ядер расщепляются на два молекулярных одноэлектронных σ-уровня, один из которых соответствует молекулярной орбитали, симметричной относительной плоскости, равноотстоящей от ядер и перпендикулярной к оси молекулы. Этот уровень, согласно Хунду (а также Гайтлеру и Лондону), лежит ниже, чем второй σu-уровень, соответствующий антисимметричной орбитали.
При мысленном сведении ядер до их слияния симметричная молекулярная оуорбиталь переходит в 1s-орбиталь объединенного атома, антисимметричная - в 2р-орбиталь. Поэтому эти состояния Хунд обозначает символами 1sσ и 2рσ т. е. он рассматривает молекулу с точки зрения объединенного атома. Такой взгляд был впоследствии подвергнут критике Леннард-Джонсом и Герцбергом.
Для четырех первых электронов двухатомной молекулы при большом межъядерном расстоянии реализуется конфигурация (1sσ)2(2pσ)2. Если затем добавить к ним пятый, то ему будет соответствовать 2s-орбиталь разъединенных атомов. Две таких орбитали, принадлежащие разным атомам, при сближении ядер преобразуются в симметричную и антисимметричную молекулярные σ-орбитали, причем энергия первой ниже, чем энергия второй, что следует из корреляции этих МО с орбиталями объединенного атома: симметричной МО соответствует 2sσ-AO, антисимметричной - 3pσ.
Таким образом, следует ожидать следующую последовательность одноэлектронных состояний двухатомной молекулы в порядке возрастания соответствующих им энергетических уровней: 1sσ, 2pσ, 2sσ, 3pσ, ...
Для первых восьми электронов при больших межъядерных расстояниях реализуется конфигурация (1sσ)2(2pσ)2(2sσ)2(3pσ)2. Девятый электрон соответствует 2р-орбитали разъединенного атома. Шесть таких орбиталей (по три от каждого атома) преобразуются при сближении ядер в следующие МО: симметричную σg-МО, антисимметричную σu-МО и две двукратно вырожденные симметричные πu-МО. Устанавливая соответствие между МО и АО объединенного атома, Хунд определил, что первая из названных выше МО является sσ (или dσ)-орбиталью, вторая- рσ-орбиталью, третья - pσ - и последняя dπ-орбиталями. При этом состояние 3sσ по энергии должно лежать ниже, чем 4рσ, а 2рπ ниже, чем 3dπ. По мнению Хунда, наиболее вероятной является следующая последовательность одноэлектронных состояний в порядке возрастания их энергии:
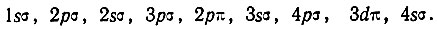
Таким образом, в 1927-1929 гг. Хундом были в качественном виде сформулированы некоторые важные идеи (одноэлектронного приближения, соответствия между атомными и молекулярными состояниями и т. п.), получившие затем более глубокую разработку. Однако его рассуждения о природе химической связи не являются специфическими для метода молекулярных орбиталей, а соответствуют более общему уровню рассмотрения, на котором не проявляются различия методов ВС и МО.
Другим исследователем, внесшим большой вклад в развитие молекулярно-орбитальной теории, был американский ученый Малликен. В 1925 г., изучая закономерности в молекулярных спектрах и сопоставляя их с атомными, он отметил сходство в спектральных характеристиках молекул CN, CO+, N2+, ВО, BeF со спектром Na. Подобные аналоги были установлены в 1925-1927 гг. в работах Мекке, Бэрджа, Шпонер и других на примере молекул СО и N2 и атома Mg, молекулы NO и атома Аl и т. п. Так, сопоставляя структуру молекулярных и атомных спектров, Бэрдж предположил, что энергетические уровни, связанные с валентными электронами молекулы, соответствуют "во всех существенных аспектах", т. е. по характеру вырождения, мультиплетности и взаимному расположению на энергетической шкале, уровням, на которых находятся валентные электроны в изоэлектронных, точнее изовалентноэлектронных, атомах. По предложению Бэрджа, молекулярные уровни стали обозначаться теми же буквами (s, p, d, f,... и т. п.), что и атомные, но только заглавными. Его обозначения 1S, 1Р, lD, 2S, 2Р соответствуют современным: 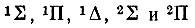 (с подуровнями 2∏1/2 и 2∏3/2).
(с подуровнями 2∏1/2 и 2∏3/2).
Указанные аналогии натолкнули Малликена на мысль, что каждому электрону в молекуле можно приписать определенную орбиту [64-65]. Например, электроны в молекулах CN и ВО должны характеризоваться квантовыми числами, аналогичными квантовым числам в атоме Na (хотя эти молекулы имеют на два K-электрона больше). Созданная Малликеном теория в значительной степени основана на изложенных выше идеях Хунда. Малликен отмечает, что интерполяция между случаями строгб разделенных атомов и объединенного атома, проводившаяся Хундом, оказывается полезной для оценки электронного состояния двухатомных молекул. В частности, модель объединенного атома позволяет использовать принцип Паули для определения максимально возможного числа электронов, соответствующих любым заданным квантовым числам. Квантовые числа, характеризующие электронное состояние молекулы, получаются из квантовых чисел, соответствующих электронному состоянию объединенного атома в предположении, что этот атом помещен в сильное электрическое поле. Наложение последнего эквивалентно мысленному расщеплению ядра объединенного атома на отдельные ядра, входящие в молекулу.
Однако реальная последовательность термов по энергии может отличаться (и весьма значительно!) от последовательности, имеющей место в сильном электрическом поле. Распределение электронов для основного состояния молекулы может соответствовать их распределению в некотором возбужденном состоянии объединенного атома, и наоборот.
Так как основная часть информации о прочности химических связей основана на спектроскопических данных, Малликен высказал предположение, что при анализе электронной структуры молекулы может оказаться полезным метод, аналогичный использованному Бором для определения электронной конфигурации атомов. Этот метод состоит в том, что все электроны мысленно удаляются из атома, а затем по одиночке возвращаются в атом, занимая доступные орбиты с наиболее низкой энергией. Конечно, применение этого метода к молекулам затруднялось тем, что отсутствовала достаточная информация об энергетической последовательности орбит в молекуле. Для решения этой проблемы были использованы корреляции между предельными случаями объединенного и изолированного атомов. Развитие метода Хунда Малликеном, по мнению последнего, "состояло прежде всего в попытке определить квантовые числа отдельных электронов" [65, с. 190]. При понижении сферической симметрии изолированного атома до аксиальной электроны, характеризующиеся одними и теми же квантовыми числами n и l, но различными |m|*, уже не будут эквивалентными. Их энергия теперь зависит также от абсолютной величины квантового числа m. Таким образом, атомная оболочка ns не расщепляется, в то время как оболочки np,nd,... расщепляются на две, три,... оболочек. Одна из них (σ-типа) характеризуется нулевым значением проекции одноэлектронного момента импульса на ось квантования. Она может заполняться не более чем двумя электронами с противоположными спинами. Каждой из остальных оболочек (π-, σ-...типов) соответствуют два не нулевых, равных по абсолютной величине, но различающихся знаком, значения проекции момента импульса. Соответственно эти оболочки могут заполняться не более чем четырьмя электронами.
* (В обозначении Малликена - σl)
Таким образом, как было показано Малликеном, электронные оболочки в молекуле определяются бблыпим набором квантовых чисел, а их максимальная заселенность электронами понижается. Если замкнутые оболочки в атоме содержат 2, 6, 10,... электронов, то в линейной молекуле они содержат либо 2, либо 4 электрона.
Электронные оболочки и соответствующие им одноэлектрон-ные энергетические уровни Малликен классифицировал на связывающие (bonding) и несвязывающие (unbonding). Под связывающим он понимал такой уровень, удаление электрона с которого приводит к ослаблению химической связи, в отличие от случая удаления электрона с несвязывающего уровня*, В качестве критерия прочности связи он использовал три экспериментально наблюдаемые величины: энергию диссоциации (D), равновесное межъядерное расстояние (R0) и частоту колебания (ω0) связи двухатомной молекулы. Укорочению химической связи должно, по мнению Малликена, соответствовать увеличение D и ω0.
* (Более строго определить связывающий уровень следовало бы как уровень, удаление с которого всех электронов приводит к ослаблению связи. Так, например, для молекул М2, где М - атом щелочного металла, удаление одного электрона с верхнего, дважды занятого энергетического уровня приводит к упрочению химической связи. В этом смысле уровень не должен быть, по Малликену, связывающим. Однако удаление обоих электронов приводит к распаду молекулы (точнее, молекулярного иона), и это свидетельствует о том, что именно электроны данного уровня обеспечивают химическую связь в молекулах М2.)
Наиболее сложной проблемой было определение энергетической последовательности одноэлектронных состояний в молекуле. Поскольку о теоретическом расчете в то время не могло быть и речи, то Малликену пришлось использовать различную информацию (в основном экспериментальную): потенциалы ионизации, энергии электронных переходов и их интенсивности, эмпирически установленные правила отбора и т. п. Кроме того, он ввел дополнительное предположение о том, что число σ-, π-, δ-...электронов при переходе от объединенного атома к разделенным не изменяется. Однако такой переход неоднозначен, во-первых, потому, что в процессе разъединения могут получиться атомы в различных состояниях, а во-вторых, потому что объединенный атом может "расщепляться различными спобами" (например, 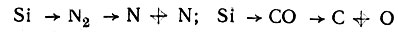 и т. д.).
и т. д.).
Ввиду неоднозначности указанного перехода существенным является использование принципа изоэлектронности, а также предположения о сохранении числа σ-, π-... электронов.
Заканчивая обсуждение работ Хунда и Малликена, остановимся на их оценке. Прежде всего следует отметить, что Хундом и Малликеном была дана вполне удовлетворительная интерпретация молекулярных спектров на основе квантовой механики. Была прослежена связь молекулярных спектров с атомными. Вместе с тем был достигнут прогресс в понимании квантовомеханической природы валентности атомов и удалось объяснить некоторые особенности их химического поведения. Существенным элементом теории Хунда-Малликена была идея одноэлектронного приближения в ее простейшей формулировке. Все эти результаты составляют непреходящую ценность их работ.
В то же время следует отметить, что метод МО* выступает р их исследованиях преимущественно как качественная полу-^мпирическая теория молекулярных спектров. Описание молекул велось в терминах энергий и мультиплетностей термов, а не волновых функций, которые определяют распределение электронной плотности в системе. Изучались фактически корреляционные диаграммы "термы разделенных атомов - термы объединенного атома", но не детальное изменение энергии отдельных термов (или одноэлектронных состояний) при изменении межъядерного расстояния, характеризующееся потенциальными кривыми взаимодействия атомов.
* (Термин "орбиталь" впервые появился, по-видимому, в 1932 г., в работе Малликена [66]. Сначала же Хунд использовал термин "bahn", a Малликен - "orbit".)
Иными словами, речь шла об отношении величин, а не о функциональном отношении Е = E(R). Особенность функциональной зависимости состоит в том, что она может включать "особые точки" (например, экстремумы), в которых объект переходит в особое состояние и происходит образование нового качества.
Для того чтобы расширить значение метода Хунда-Малликена, необходимо было прежде всего выйти за рамки чисто качественных полуэмпирических построений и найти способ количественного теоретического расчета. Новой постановке задачи способствовали работы Леннард-Джонса и Герцберга.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'