Определение содержания и выделение суммы редкоземельных элементов из руд, пород и минералов (Е.М. Гельман, К.И. Титомирова)
Для определения суммы рзэ в рудах, породах, минералах, концентратах и других материалах применяют весовые [1-4], комплексонометрические [5, 6] и фотометрические [4, 7, 8] методы анализа. Для выделения суммы рзэ используют несколько вариантов, основанных на сочетании реакций осаждения гидроокисей, фторидов и оксалатов с кальцием в качестве коллектора.
Опыт массовой работы по использованию фотометрического [7] и весового [1] методов определения малых содержаний рзэ показал, что указанные методы не обеспечивают необходимой точности и чувствительности определения. Как установлено [9-13], ошибки связаны с недостаточной полнотой выделения оксалатов рзэ, их неточным фотометрическим определением в присутствии мешающих примесей, которые не полностью отделяются в ходе анализа, особенно железа, остающегося в небольших количествах даже при самой тщательной очистке в выделенном осадке оксалатов рзэ. Влияние Fe(II) и Fe(III) сказывается в том, что железо образует в присутствии арсеназо I и салицилата натрия окрашенное соединение.
Проведенные исследования показали, что при использовании фосфата натрия для маскирования рзэ можно при фотометрировании вычесть оптическую плотность окрашенного соединения железа. Ошибки, связанные с некоторой зависимостью оптической плотности соединения рзэ с арсеназо I от содержания железа, устраняют путем построения калибровочных графиков в присутствии различных концентраций железа. Практически оказывается достаточным двух концентраций: 25 и 75 мкг Fe/25 мл. Для получения точных результатов необходимо также учесть, что соединения элементов иттриевой подгруппы с арсеназо I (и с ксиленоловым оранжевым) при большом содержании иттрия отличаются значительно более интенсивной окраской по сравнению с элементами цериевой подгруппы. При расчете содержания следует пользоваться калибровочными графиками соответствующей смеси рзэ, либо, если состав смеси рзэ анализируемых образцов не известен, средними результатами калибровочных графиков иттриевой и цериевой подгрупп.
Нами установлено, что полнота выделения оксалатов рзэ достигается при соблюдении следующих условий: концентрация рзэ в осаждаемом растворе сохраняется в пределах 0,5-7 мг/мл; величина pH раствора (устанавливают уротропином) должна быть равной 4-4,5; осадителем служит 5-20-кратный избыток насыщенного раствора Н2С2О4; осаждение проводят на холоду без коллектора; железо связывают в комплекс трилоном Б [14]. В некоторых случаях общий объем раствора составляет всего лишь несколько капель.
Метод может быть применен как для фотометрического, так и для весового определения.
Относительная ошибка весового определения ± 1-5%; фотометрического определения ± 5-15%.
Чувствительность фотометрического метода 5 × 10-3%.
0,05-0,1 г анализируемого материала в корундовом или никелевом тигле сплавляют с 1 г NaOH и 0,5 г Na2O2 при 600-700° С. Плав растворяют в 80-100 мл воды, кипятят ∼ 20 мин., остаток отфильтровывают, промывают слабым раствором NaOH и растворяют в горячей НС1 (1 : 1). Прибавляют NH4OH До запаха и еще 3-4 мл избытка. Осадок отфильтровывают* и промывают 2%-ным раствором NH4NO3. Промытый осадок сразу переносят в платиновую чашку и обрабатывают при нагревании 5-8 мл HF до получения влажных солей. Остаток растворяют в 50-60 мл горячей воды, прибавляют около 1 мл HF и 6-8 капель НС1 (для увеличения растворимости фторида железа). Фториды рзэ отфильтровывают и тщательно промывают 1%-ным раствором HF и НСl (по объему). Осадок с фильтра смывают горячей водой обратно в платиновую чашку, а фильтры озоляют и сжигают в платиновых тиглях, предварительно смочив их НС1. Полученную золу присоединяют к осадку в чашке, прибавляют 4-5 мл НС1 и выпаривают на плитке досуха. Сухой остаток обрабатывают при нагревании 2-4 мл 50-60%-ной НСlO4, 1 мл НС1 и 3-6 мл горячей воды [15], споласкивая ею стенки чашки. Кислоты выпаривают до появления белых паров HClO4, уменьшают температуру и выпаривают досуха. Сухой остаток снова обрабатывают 1-2 мл НС1 и 3-5 мл горячей воды и выпаривают досуха. Полученный остаток растворяют в 4-5 мл горячей НС1 (1 : 1), переносят в стакан емкостью 50 мл и выпаривают до слегка влажных солей. Соли растворяют в нескольких каплях горячей воды и устанавливают объем, исходя из ожидаемой концентрации рзэ, которую ориентировочно определяют в ходе анализа по величине осадка фторидов (табл. 1).К раствору прибавляют кристаллик 1-3 капли 0,1%-ного спиртового раствора индикатора пентаметоксикрасного, по каплям- 25%-ный раствор уротропина до обесцвечивания красно-фиолетовой окраски раствора и 1-2 капли избытка. К раствору прибавляют насыщенный раствор H2C2O4 (табл. 1), осадок с раствором перемешивают не менее 2мин., затем оставляют на 2-3 часа при периодическом перемешивании (лучше на ночь), фильтруют через плотный фильтр, промывают 1%-ным раствором Н2С2O4, озоляют и прокаливают в фарфоровом тигле при 600-620° С в течение 30 мин.
* (После охлаждения. )

Таблица 1. Расчет объемов исследуемого раствора и раствора осадителя. Навеска образца для фотометрического определения 0,1 г, для весового-1 г
Фотометрическое определение выполняют при содержании 0,005-3%2Ln203. Прокаленные окислы осторожно растворяют в НСl (1 ; 1), добавляют каплю 3%-ной Н2O2 и нагревают до ее полного разложения. Раствор переносят в колбу емкостью 25 мл и доводят водой до метки. Для фотометрирования 10 мл (или меньше) раствора помещают в мерную колбу емкостью 25 мл, прибавляют несколько капель 0,1%-ного спиртового раствора индикатора пентаметоксикрасного, по каплям - 5%-ный раствор NH4OH до обесцвечивания раствора л 1 N НС1 - до возвращения едва заметной окраски раствора. К раствору прибавляют 1,5 мл 0,1%-ного раствора арсеназо, 0,3 мл 5%-ного раствора салицилата натрия, 10 мл 20%-ного раствора уротропина и воду до метки. Через 30 мин. измеряют оптическую плотность раствора Ех на фотоколориметре ФЭК-М с желтым светофильтром (λ = 573 ммк) в кювете длиной 10 мм. Раствором сравнения служит вода. По окончании измерения к раствору в колбе прибавляют 4 капли 2%-ного раствора Na2HPO4; через 20 мин. снова измеряют (как указано выше) оптическую плотность раствора Е2. Разность Е1-E2 соответствует оптической плотности рзэ с арсеназо I. По калибровочному графику для соответствующей группы рзэ и величины Е2 находят содержание рзэ. Если состав смеси рзэ анализируемых образцов не известен, берут средние показания калибровочных графиков иттриевой и цериевой подгрупп.
Калибровочные графики. Для каждой подгруппы рзэ* готовят три серии растворов, содержащих по 10; 30; 50; 70 и 90 мкг соответствующей смеси в 25 мл. В одну серию прибавляют по 25 мкг FeCl3; в другую - по 75 мкг FeCl3; в третью серию растворов железо не вводят. Затем добавляют 7-10 мл воды и продолжают, как описано для фотометрического определения рзэ в анализируемых образцах.
* ( Стандартный раствор цериевой или иттриевой групп рзэ готовят растворением Ln2O3 в НС1. Состгв цериевой подгруппы: 10—20% La2O3; 20—40% СеO2; 2—10% Рr2O3; 10—30% Nd2O3; 1—5% Sm2O3, Gd2O3 и (Y2O3). Состав цттриевой подгруппы; (0,5—5% La2O3, СеO2, Рr2O3, Nd2O3 каждого); 60—70% Y2O3, 5—10% Еr2O3 и Yb2O3.)
Весовое определение выполняют при содержании в образцах от 0,5%2Ln2O3 и при любых содержаниях, если сумму окислов в дальнейшем используют для определения индивидуальных рзэ. Для анализа 0,2-1 г материала сплавляют при 600-700° С с шестикратным количеством NaOH и 0,5 г Na2O2. Плав растворяют в 120-150 мл воды, кипятят 20-30 мин., по охлаждении фильтруют и промывают слабым раствором NaOH. Осадок гидроокисей растворяют в НС1, переосаждают аммиаком и обрабатывают при нагревании HF, не содержащей свинца, до получения влажных солей. Остаток растворяют в 50-60 мл горячей воды, добавляют 1 мл HF, 0,5 мл НС1 и по охлаждении осадок фторидов фильтруют, промывают и обрабатывают НСl и НСO4, как было описано выше. В полученном растворе (∼ 10 мл) аммиаком осаждают и, если нужно, переосаждают гидроокиси рзэ для отделения от кальция. Осадок растворяю" в нескольких миллилитрах горячей НС1 (1 : 1), выпаривают до слегка влажных солей; осаждают оксалаты рзэ, как описано выше (см. табл. 1). Осадок оксалатов рзэ и тория прокаливают во взвешенном фарфоровом тигле при 600-620° С до постоянного веса.
Метод используют для массового анализа руд, пород и минералов, содержащих 0,005-25% 2Ln2O3.
В табл. 2 и 3 приведены результаты анализа различных образцов, выполненного предлагаемым методом.
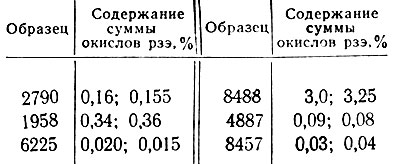
Таблица 2. Результаты фотометрического определения суммы окислов рзэ в рудах
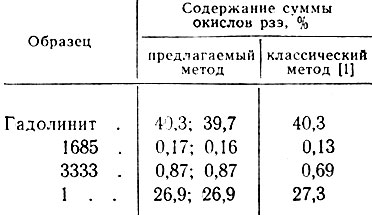
Таблица 3. Результаты весового определения суммы окислов рзэ в рудах и минералах
Литература
- Анализ минерального сырья. Под ред. Ю. Н. Книпович и Ю. В. Морачевского. Л., ГНТИ, 1956, стр. 731.
- Е. К. Корчемная. Сб.: "Методы определения и анализа редких элементов". М., Изд-во АН СССР, 1961, стр. 141.
- Ю. А. Балашов. Сб.: "Методы определения и анализа редких элементов". М., Изд-во АН СССР, 1961, стр. 142.
- В. Ф. Лукьянов, А. А. Мозжорина. Сб.: "Методы определения и анализа редких элементов". М., Изд-во АН СССР, 1961, стр. 158.
- Ю. А. Чернихов, Б. М. Добкина, Т. М. Малютина. Сб.: "Методы определения и анализа редких элементов". М., Изд-во АН СССР, 1961, стр. 170.
- G. Вrunishоlz, R. Сahen. Helv. chim. acta, 39, 324, 2136 (1956).
- Ф. В. Зайковский, В. С. Башмакова. Сб.: "Методы определения и анализа редких элементов". М., Изд-во АН СССР, 1961, стр. 156.
- В. И. Кузнецов. ЖАХ, 7, 226 (1952).
- К. Вrоadhеad, Н. Неadу. Anal. Chem., 32, 1603 (1960).
- И. П. Алимарин, Ф. И. Павлоцкая. Сб.: "Редкоземельные элементы". М.г Изд-во АН СССР, 1956, стр. 162.
- Е. М. Гельман. Отчет СЗГУ. Л., 1961.
- Ю. Н. Книпович, О. П. Бояришнова. Сб.: "Геохимия", № 6, Материалы ВСЕГЕИ, Л., 1947.
- В. Серебренников. Уч. зап. Томск, гос. ун-та, № 8, 111 (1948).
- Материалы VIII совещания работников геологических организаций, вып. 2. М., 1961.
- С. Б. Саввин, В. В. Б а грее в. Зав. лаб., 26, 412 (1960).
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'