Органические катализаторы
В современной органической химии механизмы различных реакций объясняются взаимодействием электронных оболочек реагирующих между собой атомов или молекул. При этом могут образовываться промежуточные нестабильные соединения: карбанионы, карбониевые катионы, ониевые комплексы и т. д., в какой-то степени облегчающие протекание заключительной стадии электронного взаимодействия реагентов - как с энергетической точки зрения, так и с точки зрения квантовой теории химической связи. Тогда роль органического катализатора должна сводиться к образованию с его участием чрезвычайно выгодных энергетически, наиболее вероятных в квантовомеханическом смысле промежуточных комплексов. По существу, речь идет о применении теории промежуточных соединений или так называемой теории активированного комплекса для объяснения действия органических катализаторов. Поясним сказанное на примерах.
В органической химии известна реакция кротоновой конденсации. Например, для масляного альдегида эта реакция выглядит так:
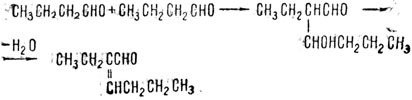
Конечный продукт носит название 2-этилгексен-2-аль-1, а промежуточный продукт - бутиральдоль. При действии органического катализатора гликокола (аминоуксусной кислоты Г) возможно получение 2-этилгексен-2-аль-1 непосредственно из масляного альдегида, минуя стадию образования бутиральдоля. Ион гликокола Г- в этой конденсации отрывает протон от масляного альдегида. При этом необходимы два условия, наличие которых требует особая специфичность данного органического катализатора: исходным веществом для кротоновой конденсации может быть только карбонильное соединение, и никакое другое с активной метиленовой группой; Г- отрывает протон от метиленовой группы масляного альдегида только при образовании промежуточного комплекса ион гликокола - альдегид. Таково специфичное действие органического катализатора в отличие от неспецифичного каталитического действия ОН- в основном катализе.
Химикам хорошо известно вещество изатин. Он является: ценным полупродуктом в производстве красителей, а также применяется для открытия примеси тиофена в бензоле в аналитической химии. В качестве органического катализатора изатин разлагает аминокислоты. Подобное действие изотина можно сравнивать с действием некоторых ферментов (дегидраз), для которых характерна следующая схема реакций:
субстрат ⋅ Н2 + катализатор ⋅ субстрат + катализатр ⋅ Н2 катализатор ⋅ Н2 + акцептор ⋅ акцептор ⋅ Н2 + катализатор
Разложение аминокислоты (α-аланина) изатином иллюстрируется суммарной реакцией
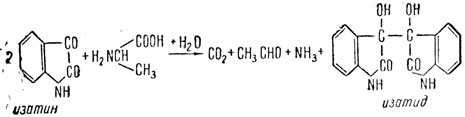
с последующей регенерацией катализатора:
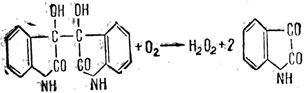
На этом примере видно, что изатин (как органический катализатор) присоединяет два атома водорода аминокислоты с образованием промежуточного вещества изатида. Водород, связанный в изатиде, более активен, чем в аминокислоте (легко реагирует с кислородом). Водород аминокислоты в таких условиях к подобной реакции с кислородом не способен. К этой реакции тесно примыкает другая каталитическая реакция с участием органического катализатора - метиленового голубого, красителя, применяемого биологами для окраски срезов ткана при изучении окислительно-восстановительных процессов в организме. Действие метиленового голубого иногда ведет к отнятию водорода от некоторых аминокислот.
Имидазол - органическое вещество, которое является составной частью витаминов B12 и B1, пуриновых оснований" входящих в состав нуклеиновых кислот, и многих других важных природных соединений. Имидазол выступает в роли органического катализатора при омылении сложных эфиоов и амидов кислот. Для того чтобы разобраться в механизме действия этого органического катализатора, нам придется привлечь некоторые элементарные понятия из современной теоретической органической химии. Так, в молекуле сложного эфира (формула которого в общем изображается так:
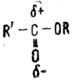
где R' и R - углеводородные или другие радикалы) δ- означает, что кислород как сильно электроотрицательный атом в какой-то степени притягивает к себе электроны углерода и поэтому несет на себе некоторый избыток электронной плотности, в то время как δ+ на углероде означает потерю доли электронной плотности. Это изменение в распределении электронной плотности между кислородом и углеродом вызвано стремлением атома кислорода дополнить свое внешнее электронное оружие до стабильного октета электронов. В имидазоле
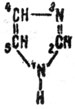
азот (3) расходует три электрона из своих пяти внешних электронов на связь с углеродами (4) и (2), а два неиспользованных электрона придают отрицательный характер этому атому азота. Поэтому понятно, почему на первой стадии реакции происходит образование промежуточного соединения (А) за счет положительного углерода эфира и отрицательного азота в имидазоле. Это промежуточное соединение неустойчиво и распадается с образованием другого промежуточного соединения (Б) достаточно устойчивого, но под действием Н2О легко омыляющегося до кислоты. При этом регенерируется катализатор - имидазол. Вот общая схема этого процесса:
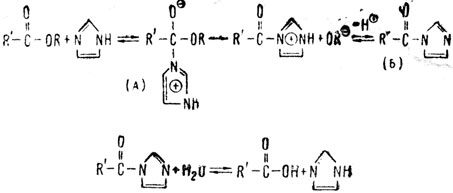
Анализируя приведенные примеры, можно сразу уловить общий признак действия органических катализаторов-перенос протона. С другой стороны, для органических катализаторов в приведенных примерах характерна специфичность действия в отличие от неспецифического действия кислотно-щелочных катализаторов. Известно, например, что действие имидазола в реакции омыления сложных эфиров специфично в высокой степени для смешанных ароматических эфиров с пара-заместителем в фенильном ядре. Явление специфичности связано с более глубокими изменениями в структуре реагирующих веществ (особенно на стадии образования промежуточных комплексов), чем это имеет место при кислотно-щелочном катализе. Здесь мы вплотную подходим к вопросу об источнике повышенной активности промежуточного комплекса в каталитических реакциях, причем в определенном направлении протекания процесса.
Согласно теории активированного комплекса любая химическая реакция должна проходить через стадию образования промежуточной критической конфигурации, соответствующей вершине активационного (или, как еще принято называть, потенциального) энергетического барьера.
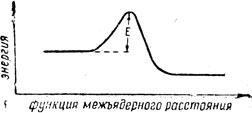
Стади промежуточной критической конфигурации
Квантовомеханическая сущность теории активированного комплекса заключается в том, что в процессе любой химической реакции пространственные координаты первоначальных конфигураций системы реагирующих атомов или молекул непрерывно изменяются, и система переходит в конечную конфигурацию через некоторое переходное состояние. Иными словами, промежуточный комплекс вещество - катализатор (или просто комплекс двух реагирующих веществ) является динамическим образованием, в котором атомы непрерывно изменяют свое положение относительно друг друга, будучи вместе с тем ограниченными в этих своих изменениях определенной структурой комплекса (конфигурацией). Это некоторое переходное состояние есть энергетически наиболее выгодный путь изменения относительных координат конфигураций атомов из начального состояния в конечное. При этом энергетический уровень промежуточного - активированного - состояния выше энергетического уровня исходного - начального - состояния.
Пусть происходит реакция между атомами А и молекулой ВС:
В молекуле ВС атомы В и С соединены простой связью с помощью двух электронов. Атом А имеет один неспаренный электрон. По мере приближения атома А к молекуле ВС взаимодействие между тремя электронами приводит к увеличению обменной энергии, и, следовательно, к увеличению потенциальной энергии. Понятие обменная энергия в квантовой химии возникает при рассмотрении взаимодействия электронов, которые могут меняться местами, переходя с орбиты одного атома на орбиты другого, взаимодействующего с первым атомом, и наоборот. Причем в силу того; что все электроны одинаковы, различить, какому атому принадлежит электрон, и определить, на какой орбите в данный момент он находится, невозможно. Таким образом, обменная энергия и есть результат этого необычного взаимодействия электронов в ходе химической реакции.
Сближение атомов сопровождается, естественно, увеличением обменной энергии. С другой стороны, электроны - отрицательные частицы, а частицы с зарядом одного знака отталкиваются друг от друга. Следовательно, при сближении электронов сила отталкивания возрастает, и возрастает их потенциальная энергия. В нашем случае увеличение потенциальной энергии обусловлено преодолением отталкивания между А и ВС за счет уменьшения притяжения между В и С. В результате система достигает такого состояния, когда между атомами А, В и С силы взаимодействия уравновешиваются. Образуется неустойчивая конфигурация А...В...С, которой и соответствует вершина потенциального барьера. Энергия, необходимая для преодоления этого барьера, есть энергия активации, которая может быть вычислена методами квантовой химии. Энергия данной трехэлектронной системы - функция межъядерного расстояния А - В и В - С, если все три атома находятся на одной прямой линии. Потенциальная энергия активированного комплекса является максимальной лишь в направлении, по которому должна протекать данная реакция, что и определяет специфичность действия активированного комплекса. Известно, что атом не неподвижная точка, а частица, которая находится в беспрерывном движении. Причем виды движения атомов в молекуле весьма разнообразны: поступательное, вращательное и колебательное. Каждый вид такого движения требует соответствующей затраты энергии. В процессе активации реагирующей системы чрезвычайно велико значение соотношения поступательной и колебательной энергии взаимодействующих атомов. Поступательная энергия, например, атома А в системе А + ВС направлена в целом на преодоление потенциального барьера в реакции, а колебательная энергия молекулы ВС является одним из источников затормаживания этого процесса. Когда мы имеем дело со второй частью процесса реакции, т. е. с разложением активированного комплекса до конечных продуктов, существенное значение имеет снятие колебательной энергии в двухатомной системе А - В с помощью третьей частицы С. Благодаря этому уменьшается возможность обратимого разложения активированного комплекса с образованием первоначальных веществ.
Теперь мы должны попытаться ответить на следующие вопросы, связанные с участием в химических реакциях органических катализаторов: на какой стадии каталитического процесса катализатор начинает активацию исходных веществ? в чем заключается активирование катализатором исходных веществ?, какова природа специфичности действия определенного катализатора на определенный реагент? В первый момент кажется бессмысленным отвечать на эти вопросы в рамках органического катализа, поскольку они затрагивают общую проблему реакционноспособности органических веществ, участвующих в каталитических или некаталитических реакциях. Тем не менее имеется существенное отличие каталитической активации от обычной активации молекул, вступающих в реакцию. Видимо, молекулы реагирующих веществ в присутствии катализаторов переходят в активное состояние еще до образования собственно активированного комплекса вещество - катализатор, где преобладают ковалентные, координационные связи, т. е. уже химические связи. Интересно в этом отношении действие тиаминпирофосфата в реакциях образования ацетона и ацетальдегида из пировиноградной кислоты. Тиамин - химическое название витамина В1 Тиамин в форме пирофосфата служит основной частью ферментов для ряда биохимических реакций, при которых расщепляются углерод-углеродные связи. Пировиноградная кислота - важное вещество, получающееся в результате сложного процесса "сгорания" углеводов (глюкозы) в живом организме. Эта кислота достаточно известна химикам. В упомянутой реакции исходное вещество - пировиноградная кислота - подвергается предварительному активированию органическим катализатором - тиаминпирофосфатом, с образованием так называемой "активной" пировиноградной кислоты
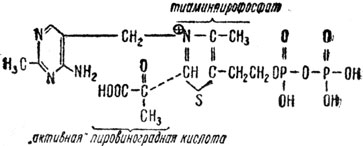
'Активная' пировиноградная кислота
После удаления карбоксильной группы из "активной" пировиноградной кислоты в виде СО2 образуется активированная форма ацетальдегида - "активный" ацетальдегид, которая в биологических системах может поставляться в виде ацетила СН3СО для образования ацетилкоэнзима А. Ацетилкоэнзим А является важнейшим ацетилирующим агентом в живых организмах. В частности, с помощью этого коэнзима вещество холил ацетилируется в ацетилхолин - химическое соединение, обеспечивающее передачу нервных импульсов.
Но вернемся к нашей реакции. После освобождения из-под влияния органического катализатора "активный" ацетальдегид превращается в обычный ацетальдегид. Образование подобных активных форм в этой реакции доказано хроматографическим анализом продуктов реакции с применением меченых атомоз С14. При этом можно предположить возникновение активированного состояния исходного вещества до преодоления потенциального барьера и сохранение активированного состояния после перехода потенциального барьера. Такие активированные состояния соответствуют определенным уровням колебательной энергии исходной системы и являются дискретными состояниями системы или, выражаясь языком квантовой химии, квантованными состояниями, для которых непрерывный переход из одного состояния в другое невозможен. Трудно еще сказать, на какой стадии каталитического процесса катализатор начинает влиять на ход активации исходного вещества, "разрыхляет" его, вызывает перераспределение энергий связей, но, видимо, на первых порах влияние катализатора носит характер не химического, а физического межмолекулярного воздействия. Это предположение обосновывается у ряда авторов, ведущих подобные исследования. Лангенбек, исследуя каталитическую активность производного уже известного нам изатина - 7-метилизатин - 4-карбоновой кислоты как модели фермента, отнимающего водород от аминокислот, считает необходимым образовав ние водородной связи как фактора, облегчающего последующий акт каталитического воздействия - перенос протона или электронной пары с образованием активированного комплекса субстрат - катализатор:
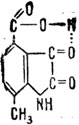
Как известно, водородная связь, образующаяся между водородом и электроотрицательным атомом типа О или N, не является валентной химической связью и обеспечивается в основном межмолекулярным физическим взаимодействием электростатического характера, возникающим в результате неравномерного распределения электронных зарядов в молекулах. В данном случае речь идет не о предварительном активировании исходного вещества, а о наличии в структуре самого органического катализатора особых предпосылок (не совсем химического порядка) для осуществления специфического каталитического действия на некоторые соединения. Следовательно, основному каталитическому действию предшествует либо предварительное активирование исходных веществ катализатором, либо активирование самого катализатора, специфичное для данного типа реакции. И то и другое предварительное активирование происходит за счет сил межмолекулярного взаимодействия. Функции, катализатора сводятся на первой стадии каталитического акта к притягиванию молекулы вещества к катализатору под действием упомянутых межмолекулярных сил. Избыточную энергию, необходимую для активации вещества, катализатор приобретает за счет целесообразности своей структуры (конкретно пря протекании данной реакции) или за счет сил межмолекулярного взаимодействия (непосредственно в процессе взаимодействия), величина которых зависит от целесообразного сочетания энергетических и структурных элементов вещества и катализатора, наличия резонансного эффекта, взаимодействия полей и т. п.
Водородная связь, как говорилось выше, также может играть активную роль в подготовке структуры самого катализатора к воздействию на субстрат или уже в ходе каталитического взаимодействия.
Специфичность каталитического действия есть результат выполнения обязательного требования, предъявляемого к катализатору и реагенту: максимальное структурное и энергетическое соответствие друг другу.
Одним из доказательств реальности только что приведенного тезиса являются работы Свейна и Брауна, в которых была исследовано действие органического катализатора 2-оксипиридина
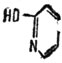
Действие 2-оксипиридина
В молекуле этого катализатора находятся одновременно дв* каталитических центра, действующих совместно, но в различных направлениях. Действие одного центра носит нуклеофильный характер, действие другого - электрофильный. Можно предположить, что нуклеофильным центром здесь будет атом азота, а электрофильным - атом углерода, связанный с гидроксильной группой. Рассматриваемый нами 2-оксипиридин - катализатор для мутаротации тетраметилглюкозы в бензольной растворе. Реакция эта несложная. Но вот что интересно в ней с точки зрения катализа. Оказалось, что если разъединить два каталитических центра в 2-оксипиридине, т. е. вместе него применить два различных катализатора - фенол и пиридин в смеси, то каталитическое действие их окажется в 7000 раз слабее, чем действие 2-оксипиридина. Отсюда ясно, какое важное значение имеют структура катализатора, расположение активных центров по отношению к структуре субстрата в данной, специфической реакции.
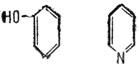
Фенол и пиридин
Читатель, наверно, уже заметил, что мы несколько раз пробовали пояснить некоторые каталитические явления с помощью квантовомеханических представлений. Это дань времени.; Сейчас каждое химическое явление принято рассматривать в трек аспектах: квантовомеханическом, термодинамическом и кинетическом. Наиболее важным из них является квантовомеханический. Этот аспект связан прежде всего с современными электронными представлениями в химии. Однако термодинамический подход к явлению катализа тоже чрезвычайно важен. К необходимости рассмотрения каталитических процессов с термодинамической точки зрения приводят два обстоятельства. Первое обстоятельство заключается в том, что катализ как явление макромолекулярное требует перехода от квантовоме-ханического толкования природы одной или нескольких микрочастиц (атома или молекулы) к статистическому описанию состояний множества частиц. Для систем, содержащих большое число молекул, так называемая квантовомеханическая статистическая сумма состояний представляет собой наиболее удобную форму, связывающую микроскопические свойства отдельных молекул с макроскопическими свойствами тел. Таким образом, речь идет о распространении квантовомеханических положений на явления в макросистемах, поведение которых регулируется термодинамикой. Второе обстоятельство, требующее рассмотрения каталитического процесса с термодинамической точки зрения, заключается в том, что температурный режим и энергетическая характеристика вещества и катализатора, энтропийный фактор - неотъемлемые параметры каталитического процесса и необходимы для управления этим процессом при решении проблемы подбора катализаторов. Хотя катализатор; не сдвигает равновесие реакции при данной температуре, т. е. каталитический процесс в конечном счете равновесный процесс, в органическом катализе (по аналогии с ферментативными системами) могут существовать несколько иные подходы к предварительным (и последующим) стадиям равновесного термодинамического состояния. Подавляющее большинство обычных химических реакций, в том числе и каталитических, протекает в условиях замкнутой системы при стремлении ее к равновесному состоянию. Протекание реакции в каком-либо направлении связано с уменьшением свободной энергии или термодинамического потенциала системы. Изменение свободной энергии является, таким образом, движущей силой реакции. Однако это изменение несамопроизвольно, а подчинено условиям равновесия данной реакции, которые объединяются общим понятием константы равновесия при данной температуре. В классической формуле расчета константы равновесия реакции
А является выражением вероятности протекания реакции в данном направлении, а множитель е-Е/RT выражает борьбу противоположных свойств (стабильности и лабильности) реагирующих веществ в зависимости от энергии активации Е и температуры Т. Изменение свободной энергии, т. е. благоприятную возможность осуществления данной реакции, можно выразить через константу равновесия данной реакции. Термодинамика указывает на вероятность осуществления реакции при данных условиях с помощью определенных термодинамических функций. Вероятностный, статистический характер термодинамических представлений как нельзя лучше сочетается с вероятностными, статистическими принципами квантовой механики. Поэтому теперь все чаще символ вероятности А в выражении для константы равновесия заменяется более полным и точным отношением Qнач/Qкон, заимствованным из квантовой химии и являющимся отношением сумм начальных и конечных состояний реагирующей молекулы. Статистическая сумма состояний реагирующей молекулы Q зависит от поступательного, вращательного и колебательного движений молекулы Q = Qпocт ⋅ Qвращ ⋅ Qколеб
Так обстоит дело с современной трактовкой протекания химической реакции в условиях равновесного процесса. В приложении к каталитическим реакциям эта трактовка выглядит следующим образом. Согласно теории активированного комплекса в процессе реакции устанавливается динамическое равновесие:
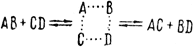
При перегруппировке связей в активированном комплексе требуется затрата определенной энергии активации. Она влечет за собой изменение энтропии и свободной энергии. Энтропия- своеобразное, но конкретное понятие о степени упорядоченности; например, переход вещества из жидкого состояния в газообразное сопровождается увеличением энтропии. В нашем случае это означает, что увеличение компактности активированного комплекса связано с уменьшением энтропии. Однако согласно законам термодинамики в окружающем нас мире энтропия или постоянна или стремится к увеличению. Поэтому уменьшение значения энтропии должно вызываться мощным противодействием ее главному стремлению. Таким мощным противодействием является стремление свободной энергии к минимуму. Все эти величины (свободная энергия, энергия активации и энтропия) связаны широко известным в термодинамике уравнением:
Значения энтропии растут при так называемом "разрыхлении" комплекса и уменьшаются при "сжатии" комплекса. Поскольку в равновесном процессе энтропия либо постоянна, либо непременно растет, "разрыхление" активированного комплекса способствует протеканию реакции в определенном направлении и, как будет сказано ниже, ведет к увеличению скорости реакции. Катализатор способствует либо "разрыхлению", либо образованию активированного комплекса, т. е. увеличению ΔS или уменьшению ΔЕ, что в конечном счете приводит к увеличению скорости реакции. В качестве иллюстрации этого положения можно рассмотреть механизм внутримолекулярного имидазольного катализа. Сложные эфиры, содержащие в своем составе имидазольное кольцо, чревычайно легко омыляются. Вот что происходит, если в качестве примера взять сложный эфир такого вида:
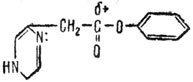
Легкость гидролиза этого эфира объясняется внутренним стерическим сжатием всей молекулы в результате каталитического процесса, сближающего реакционные центры. При этом снижается энергия активации нуклеофильной атаки неподеленною электронной парой азота положительного атома углерода (избыток положительного заряда на атоме углерода объясняется наличием сильного электроотрицательного атома кислорода карбонильной группы). Нуклеофильная атака ведет к образованию промежуточного соединения (А), и интенсивность этой стадии реакции является определяющей во всем процессе гидролиза сложного эфира:
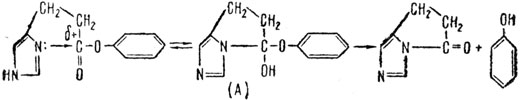
Следовательно, каталитическое действие имидазольного кольца выражается здесь в легкости образования промежуточного активированного комплекса, определяющего течение всей реакции. Кстати, такой внутримолекулярный катализ с участием органических катализаторов имеет место и во многих ферментативных процессах. Известно, что в процессе действия ферментов белковые молекулы изгибаются, сворачиваются, принимая наиболее выгодное пространственное положение, облегчающее это действие.
Итак, при действии органического катализатора можно различать преимущественное значение либо энергии активации (образование промежуточного комплекса), либо энтропийного фактора (разложение промежуточного комплекса). Иными словами, катализатор оказывает влияние либо на скорость (K1) первой стадии реакции, либо на скорость (К2) второй стадии реакции:
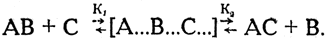
Следует также учесть, что в химической реакции энтропийный фактор состоит из двух слагаемых: изменения энтропии при обмене с внешней средой и изменения энтропии, вызванного самой химической реакцией; причем второе слагаемое энтропийной суммы зависит от степени упорядоченности исходных веществ. Изменение (упорядочение) конфигурации исходных веществ в процессе реакции при образовании переходного комплекса и дальнейшее протекание реакции в нужном направлении связаны с уменьшением энтропии реакции; катализатор же способствует некоторым наиболее рациональным для данной реакции изменениям в структуре исходных веществ еще до стадии образования промежуточного комплекса.
Если термодинамика дает возможность рассчитать вероятность протекания реакции в данном направлении, а также охарактеризовать равновесное состояние реагирующих систем, то кинетика химических реакций ставит перед собой задачу изучить механизм реакций путем сравнения скоростей (и констант скоростей) реакции на ее отдельных стадиях. Естественно, что кинетические величины тесно связаны с термодинамическими функциями. При изучении кинетики образования промежуточного комплекса нужно исходить из двух положений! 1) скорость реакции определяется скоростью "преодоления" активированным комплексом потенциального барьера; 2) исходное вещество (реагент) находится в равновесии с активированным комплексом.
Процесс преодоления активированным комплексом потенциального барьера можно моделировать процессом прохождения материальной точки через потенциальный барьер. С точки зрения квантовомеханических представлений любое передвижение материальной точки характеризуется пространственными координатами и моментами количества движения. С другой стороны, чтобы вычислить среднюю скорость материальной точки, отождествленной с молекулой, необходимо знать для всех частиц, участвующих в реакции, их статистическое распределение по энергии. Эти предпосылки легли в основу теории абсолютных скоростей реакции, уравнения которой и позволяют не только рассчитать среднюю скорость материальной точки, но и обобщить расчеты на случай множества материальных точек в единице объема. Вывод главного уравнения теории абсолютных скоростей (как и само уравнение) достаточно сложен, и мы лишены возможности изобразить его на этих страницах. Вывод его квантовомеханическим методом предусматривает наличие между исходными и конечными веществами реакции целого ряда промежуточных конфигураций активированных молекул, находящихся между собой в равновесных состояниях.
Первоначальную конфигурацию активированной молекулы нельзя отождествлять с активным комплексом, соответствующим вершине потенциального барьера. Расстояние между атомами молекулы, имеющей первоначальную конфигурацию, несколько больше, чем в случае активированного комплекса, но геометрическая форма первоначальной активной конфигурация молекулы должна быть уже идентична форме активированного комплекса, соответствующего вершине барьера.
Значение теории абсолютных скоростей заключается еще и в том, что она позволила связать кинетические факторы с термодинамическими и подтвердила зависимость ускорения реакции прежде всего от свободной энергии активации. Любой внешний фактор, уменьшающий свободную энергию активации, может ускорить реакцию. К числу этих факторов относится действие катализаторов. Причем, как уже говорилось, катализатор может действовать либо за счет изменения энергии активации, либо за счет изменения энтропии, поскольку свободная энергия активации слагается именно из этих величин. Большинство реакций гетерогенного катализа ускоряется за счет изменения энергии активации. Наоборот, многие ферментативные реакции ускоряются за счет изменения энтропии активации. Изучение кинетики некоторых реакций с участием органических катализаторов позволяет сделать довольно убедительные предположения о механизме этих реакций и предсказать оптимальные условия их проведения. Так, органические катализаторы ацетилхинин, ацетилхинидин и бруцин - представители класса алкалоидов - были успешно применены в тонком органическом синтезе, где конечные продукты в зависимости от условий реакции отличались друг от друга только различным пространственным расположением одних и тех же функциональных групп в молекуле. В химии этот вид синтеза известен как асимметрический синтез.
Экспериментально была показана стереоспецифичность реакции в случае только температурных воздействий. С помощью кинетического метода было установлено, что в присутствии катализатора при низких температурах (от -70 до 0°С) образуется один стереоизомер. При этом, как оказалось, скорость всей реакции определялась скоростью образования промежуточного комплекса. При дальнейшем повышении температуры в интервале от +13 до +57°С в основном получается другой стереоизомер. В этом случае скорость всей реакции зависела от скорости распада промежуточного комплекса. Не касаясь здесь вопроса о зависимости стереоспецифичности реакции от температуры и природы катализатора, можно сказать, что правильно примененные кинетические данные позволили выяснить приближенный механизм реакции.
Другой пример. В реакции гидролиза смешанных сложных эфиров было обнаружено каталитическое действие пирокатехина. Причем уменьшение или увеличение щелочности среды не влияло на скорость гидролиза. Кинетическим методом было выяснено, что концентрация ионов ОН- не имеет отношения к стадии реакции, определяющей скорость всей реакции в целом. Скорость всей реакции определялась скоростью образования промежуточного комплекса сложный эфир - катализатор. И здесь кинетические исследования позволили сделать вывод об обязательном протекании реакции через промежуточное соединение субстрат - катализатор и о том, что скорость образования промежуточного соединения определяет общую скорость реакции.
Перспективы применения органических катализаторов в недалеком будущем весьма обширны. Во-первых, поиск и синтез органических веществ, которые по своим каталитическим свойствам могли бы значительно превосходить действие природных ферментов. Такие вещества уже имеются и сейчас. В окислительно-восстановительных процессах, связанных с дыханием человека, большую роль играют ферменты типа витамина В2. Ученым удалось получить 2-окси-3-аминофеназин - вещество с аналогичным каталитическим действием. Органические катализаторы - алкалоиды (никотин, хинин, хинидин) в определенных условиях воспроизводят действие фермента карбоксилазы, бруцин, стрихнин - действие фермента эстеразы. Синтетические аналоги ферментов могут оказать и оказывают неоценимую помощь в изучении механизма действия ферментов и зачастую сами обладают большой физиологической активностью. Другим важнейшим направлением в области практического применения органических катализаторов является использование их в тонком органическом синтезе - как промышленном, так и препаративном. Например, в качестве мягкого дегидрирующего катализатора уже в настоящее время в некоторых реакциях применяют нитробензол. В производстве красителей долгое время широко применялся и применяется в некоторых случаях и сейчас (в качестве восстановителя) органический катализатор антрахинон. Органические смолы в виде так называемых ионитов, употребляемых для разделения и очистки различных веществ, могут катализировать отдельные реакции. Сульфофенолформальдегидные и сульфополистирольные смолы катализируют реакцию этерификации, реакцию получения (из этилового спирта и окиси этилена) ценного промышленного растворителя - этилцеллозольва; некоторые органические смолы в качестве катализаторов ускоряют полимеризацию изопреновых углеводородов. Этот процесс представляет особый интерес, поскольку промышленная полимеризация изопрена ведет к получению синтетического каучука. Наконец, представляется очень заманчивым использование органических катализаторов во многокомпонентных каталитических системах, обеспечивающих саморегулирование процесса производства сложных органических продуктов. Интересным примером такого приведения органически катализаторов (правда, пока в лабораторных масштабах) является применение ароматических кетонов для восстановления ароматических азотсодержащих соединений. Суть этой реакции в следующем.
Каждому химику знакома реакция Н. Н. Зинина - получение анилина путем восстановления нитробензола водородом. Как было выяснено, во время этой реакции образуются различные промежуточные вещества, особенно если реакция ведется в щелочной среде. Большинство этих промежуточных веществ - продуктов неполного восстановления нитробензола - могут быть выделены (используются во многих других синтезах). В процессе, о котором мы собираемся рассказать, вместо нитробензола берется похожий на него Ο-нитротолуол, а в качестве катализатора довольно сложный кетон - флюоренон. В реакционной смеси образуется несколько равновесных систем, которые взаимодействуют между собой. Действующим началом в этом взаимодействии является органический катализатор флюоренон. При этом карбонильная группа этого кетона в процессе реакции может восстанавливаться до гидроксильной, и тогда кетон (флюоренон) превращается в соответствующий спирт - флюоренол. Это первая лабильная равновесная окислительно-восстановительная система в описываемой реакции. Последующие окислительно-восстановительные равновесные системы образуются при взаимодействии системы флюоренон ↔ флюоренол с Ο-нитротолуолом и промежуточными продуктами его восстановления. Во время взаимодействия этих систем водород как бы "скользит" от одного вещества к другому, полнее восстанавливая Ο-нитротолуол. Источником водорода является раствор NaOH в метиловом спирте. Все дело заключается в том, что флюоренон гораздо легче восстанавливается в этой среде до флюоренола, чем о-нитротолуол. Но флюоренол легко передает свой водород, полученный при восстановлении, более инертному в этих условиях о-нитробензолу. Система флюоренон ↔ флюоренол снижает активационный барьер в реакции восстановления 0-нитробензола в щелочной среде, т. е. действует как типичный катализатор.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'