Каталитические свойства окислов редкоземельных элементов (А. А. Толсшопятова)
Возможность использования рзэ и их соединений в качестве катализаторов изучена недостаточно. Однако имеющиеся немногочисленные литературные данные показывают, что рзэ, особенно их окислы, могут служить катализаторами для разнообразных органических реакций. Изучение каталитических свойств рзэ представляет интерес, в частности, и для разработки теории подбора катализаторов.
Для предварительного вычисления [1] каталитической активности предложено использовать энергию связей твердых тел с реагирующими атомами органических молекул. Поэтому нами систематически определялись энергии связей кинетическим методом [2] для рзэ как продолжение исследований о зависимости каталитических свойств элементов от их положения в периодической системе [1,3-8].
Нами была изучена кинетика реакций дегидрогенизации и дегидратации этилового, н. пропилового, изопропилового и изобутилового спиртов и дегидрогенизации тетралина на окислах La, Pr, Nd, Sm, Gd, Dy, Но, Er, Tu и Yb.
Были определены энергии активации указанных реакций, относительные адсорбционные коэффициенты продуктов реакции дегидрогенизации и дегидратации изопропилового спирта (водорода, ацетона, пропилена и воды) и относительные адсорбционные коэффициенты продуктов реакции дегидрогенизации тетралина (водорода и нафталина). Рассчитаны термодинамические функции: изменение свободной энергии ΔF, тепло-содержания ΔН и энтропии ΔS процессов адсорбционного вытеснения исходного вещества продуктами реакции на каталитически активной поверхности. Определены энергии связи атомов С, Н и О реагирующих молекул с активной поверхностью катализаторов.
Для приготовления катализаторов окислы La, Pr, Nd, Sm, Gd, Dy, Но, Er, Tu и Yb, прокаленные при 800° С, были растворены в HNO3 (х. ч.) (уд. в. 1,16,-1,20) и осаждены 20%-ным раствором NH4OH. Осадки гидроокисей промывали декантацией несколько раз, отфильтровывали и высушивали при 120° С. Гидроокиси дегидратировали при постепенном повышении температуры до 600° С в токе воздуха, затем в течение часа - при 600° С. Исходная окись лантана была приготовлена из двух образцов окиси, содержащей 99,5 и 98,8% La2O3 (катализаторы La2O3 - № 1 и 2). Кроме того, окись лантана готовили еще из двойного нитрата лантана и аммония (катализатор La2O3 - № 3). Гидроокись осаждали аммиаком и обрабатывали, как указано выше.
Аппаратура и методика работы
Реакции проводили в каталитической установке проточного типа с автоматической подачей исходных жидких веществ. Реактором служила кварцевая трубка. Жидкие продукты реакции собирали в охлаждаемом приемнике, газообразные - в автоматическом газометре системы Патрикеева. Температуру измеряли хромель-алюмелевой термопарой,конец которой находился в кармане в середине слоя катализатора. Газ анализировали посредством аппарата ВТИ, а также хромотографическим методом. Катализаторы между опытами регенерировали в течение часа в; токе воздуха.
Кинетические опыты проводили так, чтобы процент превращения исходного вещества не превышал 30. Ввиду небольшого процента превращения можно порядок реакции приближенно считать нулевым и находить энергии активации Е по уравнению Аррениуса из температурной зависимости скорости выделения водорода m2 (в мл НТД/мин) и при дегидратации на основании температурной зависимости скорости образования непредельных углеводородов mCnH2n или mH2O (в мл НТД/мин). Все опыты проводили в кинетической области. Термодинамический расчет констант равновесия и равновесных степеней превращения показал, что реакции дегидрогенизации и дегидратации спиртов и дегидрогенизации углеводородов изучали в условиях, далеких от равновесия.
Результаты, полученные для всех исследованных нами рзэ, приведены в табл. 1. Найденные энергии активации для дегидрогенизации тетралина обозначены Е1, для дегидрогенизации изопропилового спирта - E2 и для дегидратации изопропилового спирта - Е3. Энергии связи Н, С и О с катализатором К рассчитывали по формулам, вытекающим из мультиплетной теории [2]:
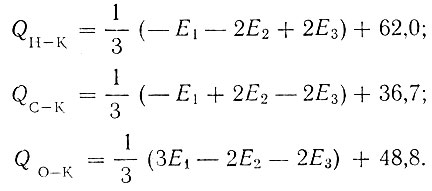
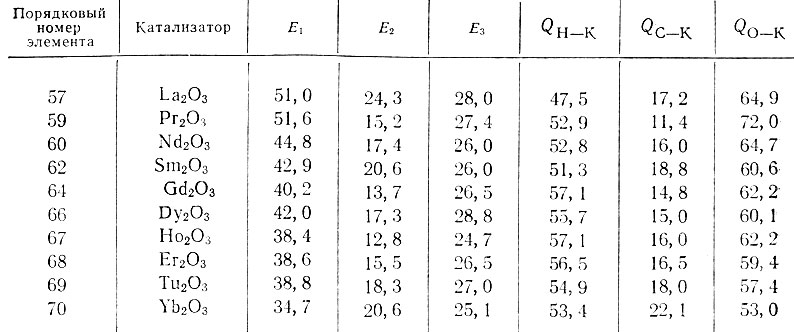
Таблица 1. Энергии активации Е (в ккал/моль) и энергии связи Q (в ккал/моль) для редкоземельных катализаторов
Для энергий связи С - Н, С - О и С - С были использованы данные Кондратьева [9, 10]: 98,1; 85,5 и 110,8 ккал соответственно.
Было найдено, что для различных редкоземельных катализаторов энергии связей реагирующих атомов QH-K, QC-K и QO-K заметно различаются.
Оказывается, что энергия связей QH-K, QC-K и QO-K -периодические функции. Наблюдается, что Q изменяется параллельно эффективному магнитному моменту μ. В последнем, как известно, периодические
свойства рзэ проявляются особенно ясно. Для QH-K этот параллелизм виден из рис. 1, а, на котором по оси абсцисс отложены атомные номера, а по оси ординат - значения QH-K из табл. 1 и значения [х из литературных данных [11]. Для QO-K наблюдается аналогичная, но менее четкая закономерность (рис. 1, б). Возможно, разброс точек происходит оттого, что, как показывают предыдущие исследования [4] с другими окислами QO-K всегда оказывается гораздо более чувствительной к различным посторонним влияниям, чем QH-K и QC-K. Энергии связи QC-K изменяются антибатно μ (рис. 2).
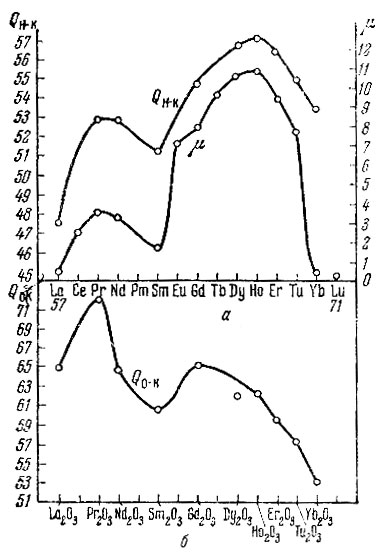
Рис.1. a-QH-K (в ккал) и μ (в магнетонах и Бора) как функция атомного номера элемента; б - QC-K(в ккал) как функция атомного номера элемента
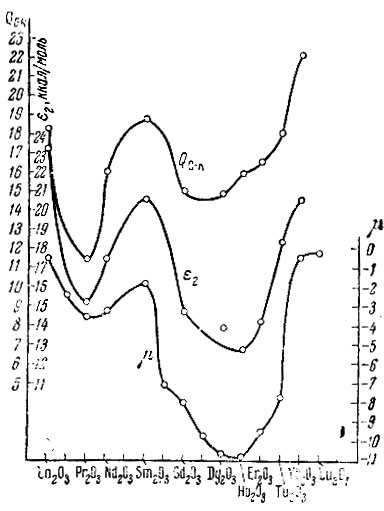
Рис. 2. QC-K (в ккал), Е2 (в ккал/моль) и μ (в магнетонах и Бора) как функция атомного номера элемента
Обратимся к изменению энергии активации Е. Энергия активации дегидрогенизации изопропилового спирта Е2 изменяется периодически и антибатно μ, подобно QC-K (см. рис. 2). Однако изменение энергии активации дегидрогенизации тетралина Е1 с изменением атомного номера элемента происходит иначе, приближаясь к непрерывному типу и изменяясь симбатно, например ионному радиусу r, рис. 3 (значения взяты из литературы [11, 12]). Энергия активации дегидратации спирта Е3 наиболее постоянна; она постепенно убывает (от 28,0 ккал для La до 25,1 ккал для Yb с разбросом точек в конце ряда, см. табл. 1).
Обнаруженные закономерности показывают, что энергии связей, зависящие от электронной структуры, представляют для катализа более первичные свойства, чем энергии активации. Действительно, все три величины - QH-K , QC-K и QO-K -закономерно периодически изменяются и симбатны или антибатны одной и той же величине μ, тогда как Е2 изменяется периодически и антибатно μ, а Е1 и Е3 изменяются непрерывно с некоторыми отклонениями. Если бы более первичным свойством было Е, то было бы непонятно, почему, например, при дегидрогенизации спирта Е2 изменяется периодически с изменением атомного номера элемента, а при аналогичной реакции дегидрогенизации тетралина Е1 постепенно падает.
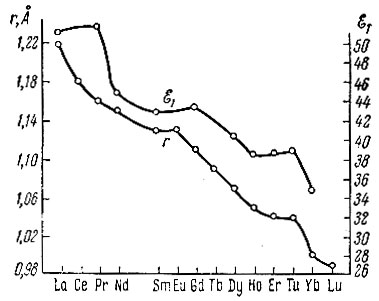
Рис. 3. E1 (в ккал/моль) и r (в А) как функция атомного номера элемента
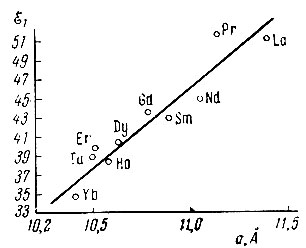
Рис. 4. Е1 (в ккал/моль) как функция а (в А)
Интересно подойти к полученным результатам с точки зрения структуры окислов рзэ. Большинство последних кристаллизуется [13] в сложной кубической решетке D53, ячейка которой содержит 16 молекул. Параметр решетки а (ребро элементарного куба) пропорционален d - наименьшему межатомному расстоянию Me - Me. Приближенная линейная зависимость между энергией активации дегидрогенизации тетралина Е1 и их параметром решетки а наблюдается у рзэ структуры D53 (рис. 4). Наблюдалась аналогичная линейная зависимость энергии активации дегидрогенизации изопропилового спирта на Со, Ni, Си, Pd, Ag, Pt от d [14] .
Эти закономерности подтверждают правильность энергетических уравнений мультиплетной теории [1] и основанного на ней принципа сохранения валентного угла, а также электронной теории катализа [15-18].
Полученные результаты показывают, что электронная структура глубинного f-слоя влияет на внешние валентные электроны 5d и 6s2, изменяя обусловливаемое последними химическое свойство элементов - энергию их связи с атомами реагирующих молекул. Таким образом, изменение электронной структуры, рассматриваемой в электронной теории, влияет на катализ путем изменения энергий связей с катализатором, рассматриваемых в мультиплетной теории.
Энергии активации реакций дегидрогенизации и дегидратации спиртов, дегидрогенизации углеводородов и избирательность действия катализаторов
На окислах La, Pr, Nd, Sm и Ек этиловый, н. пропиловый, изопропиловый, изобутиловый и н. бутиловый спирты претерпевают дегидрогенизацию и дегидратацию. На Ег203 изобутиловый спирт подвергается только дегидрогенизации. Третичный бутиловый спирт почти на всех катализаторах крекируется, исключение составляет La2O3 № 3, на котором происходит дегидратация третичного бутилового спирта. Об избирательности действия катализаторов можно судить по отношению
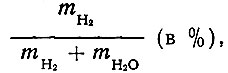
(в %). т. е. скорости образования водорода mH2 к сумме скоростей образования олефина mH2O и водорода.
Из табл. 2 и рис. 5 видно, что Рr2O3 и Sm2O3 - более дегидрирующие катализаторы, чем Lа2O3 и Nd2O3. Изобутиловый спирт на Еr2O3 только дегидрируется, а изопропиловый спирт в основном дегидратируется. Для всех исследованных спиртов и углеводородов хорошо выполняется линейная зависимость lg К от 1/7 в соответствии с уравнением Аррениуса. Наиболее легко реакции дегидрогенизации и дегидратации протекают с изопропиловым спиртом. В этом случае EH2 и EH2O - наименьшие. В ряду La2O3, Рr2O3, Nd2O3,Sm2O3 и Еr2O3 энергии активации дегидрогенизации изопропилового спирта изменяются от 14,5 до 24,3 ккал/моль, а энергии активации дегидратации - от 26,0 до 28,0 ккал/моль, т. е. EH2O изменяется всего лишь на 2 ккал/моль. Энергии активации ЕH2 и ЕH2O первичных спиртов (этиловый, н. пропиловый, изобутиловый и н. бутиловый) на всех катализаторах выше (изменяются в интервале 20-31 ккал/моль для реакции дегидрогенизации и 27-33 ккал/моль для дегидратации). Для изобутилового спирта на Рr2O3 EH2 = 34,0 и EH2O = 49,8 ккал/моль. На La2O3 № 3 третичный бутиловый спирт дегидратируется еще с меньшей энергией активации (? = 11,3 ккал/моль), чем изопропиловый. Энергии активации реакции дегидрогенизации тетралина в нафталин изменяются от 38,6 до 51,0 ккал/моль, причем с увеличением порядкового номера элемента энергии активации дегидрогенизации тетралина изменяются симбатно изменению ионного радиуса металла. Для всех веществ, исследованных в реакции дегидрогенизации и дегидратации спиртов и дегидрогенизации углеводородов на катализаторах La2O3, Pr2O3, Nd2O3, Sm2O3, Еr2O3 рассчитаны предэкспоненциальные множители уравнения Аррениуса К0 и обратные величины параметра рассеяния активных центров Е / lg К0 На изученных катализаторах для всех реакций отношение Е/ lg К0 колеблется от 3,0 до 3,7. Кроме того, если величины т, Ко и lg Ко отнести к одному квадратному метру поверхности катализатора, т. е. если Ко рассчитать по удельной скорости реакции, то величины Е/lg К0 немного возрастают, но остаются довольно постоянными, изменяясь в интервале от 4,0 до 4,7.
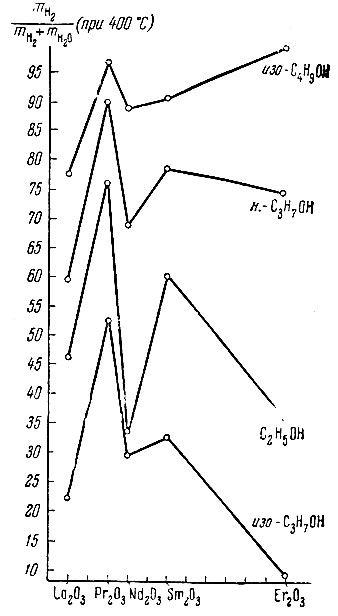
Рис. 5. Избирательность действия La2O3,Рr2О3, Nd2O3, Sm2O3 и Еr2O3
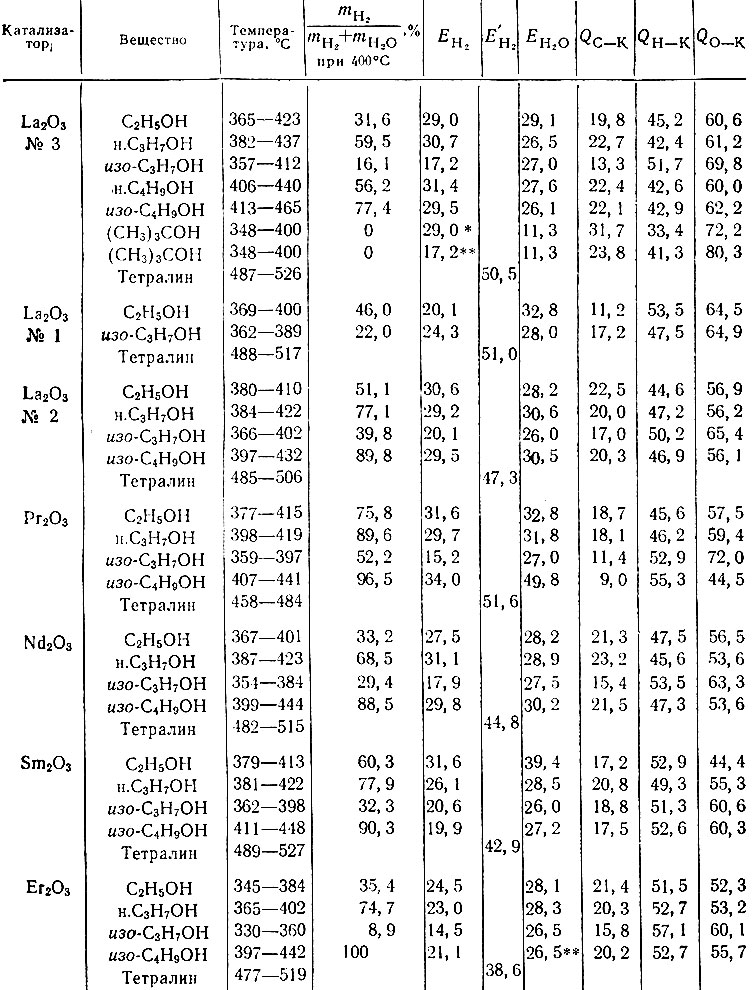
Таблица 2. Кажущиеся энергии активации реакции дегидрогенизации спиртов ЕН2 и тетралина Е'H2 , дегидратации спиртов ЕН2O на La2O3, Pr2O3, Nd2O3, Sm2O3 и Еr2O3 и энергии связей углерода QC-K, водорода QН-К и кислорода QO-K с катализаторами (в ккал/моль)
* (Энергия активации дегидрогенизации этилового спирта. )
** (Энергия активации дегидрогенизации изопропилового спирта. )
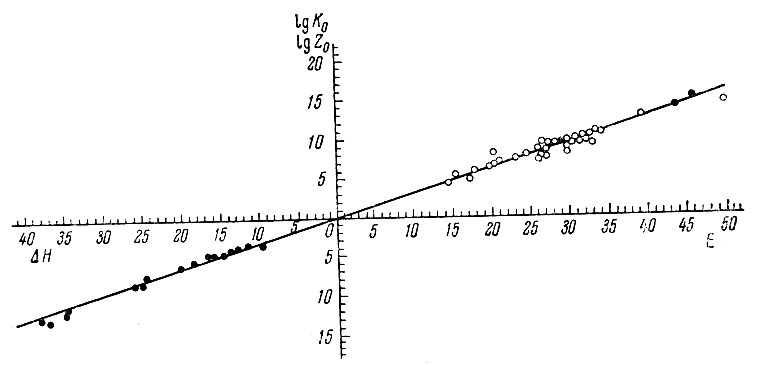
Рис. 6. Логарифмическая зависимость между К0 и Е, Z0 и ΔΗ
Таким образом, наблюдается параллелизм между изменением Е и lg К0. Из графика зависимости величины lg К0 от энергии активации Е видно, что все точки хорошо укладываются на одну прямую для всех катализаторов и для всех изученных веществ (рис. 6). Отсюда следует, что величины коэффициента распределения активных центров дегидрогенизации и дегидратации спиртов, а также дегидрогенизации углеводородов на изученных окислах близки друг к другу и что функция распределения экспоненциальна. Поскольку для разных спиртов (один и тот же катализатор) на графике зависимости величин lg К0 и Е точки укладываются на одну прямую, можно считать, что коэффициент распределения активных центров в этом случае при дегидрогенизации и дегидратации имеет одно и то же значение. Отсюда следует, что зарамочные заместители не экранируют каталитических центров.
Следовательно, между структурой реагирующей молекулы и кинетическими характеристиками наблюдается определенная связь на каждом из изученных катализаторов. Способ приготовления и химическая природа катализатора также влияют на кинетику реакций спиртов и тетралина.
Энергии связи атомов С, Н и О в реагирующих молекулах с каталитически активной поверхностью окисей лантана, празеодима, неодима, самария и эрбия
Из табл. 2 видно, что энергии связей с катализаторами La2O3, Рr2О3, Nd2O3, Sm2O3 и Еr2O3, рассчитанные по реакциям дегидрогенизации и дегидратации спиртов и дегидрогенизации тетралина, изменяются в следующем порядке:
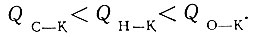
Сопоставление данных по дегидратации и дегидрогенизации первичных и вторичных спиртов различного молекулярного веса на редкоземельных катализаторах показывает, что строение спирта мало сказывается на величине энергии активации (см. табл. 1). Однако следует заметить, что дегидратация третичного бутилового спирта на La2O3 № 3 протекает легко, причем энергия активации этой реакции низкая ЕH2O = 11 ккал/моль, что, по-видимому, связано с повышенной активностью гидроксила при третичном углеродном атоме.
Близость энергий активации дегидратации для этилового, пропилового, бутилового, изобутилового спиртов на каждом из изученных катализаторов - следствие того, что молекулы спиртов ориентируются к  поверхности катализатора одной и той же группойдля дегидратации и
поверхности катализатора одной и той же группойдля дегидратации и 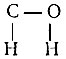 для их дегидрогенизации спиртов. Кроме того, влияние зарамочных заместителей почти в равной мере сказывается как на величинах энергий связей, разрывающихся в ходе реакции, так и на величинах возникающих энергий связей атомов реагирующих молекул с катализатором.
для их дегидрогенизации спиртов. Кроме того, влияние зарамочных заместителей почти в равной мере сказывается как на величинах энергий связей, разрывающихся в ходе реакции, так и на величинах возникающих энергий связей атомов реагирующих молекул с катализатором.
Относительные адсорбционные коэффициенты продуктов реакций изопропилового спирта и тетралина, истинные энергии активации
Для расчета истинных констант скоростей К применяли уравнение (2), вытекающее из общего кинетического уравнения Баландина [2] для мономолекулярных гетерогенно-каталитических реакций в потоке:
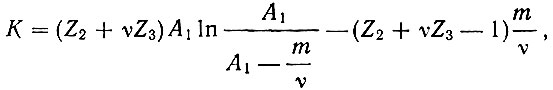 (2)
(2)где А1 скорость подачи спирта или углеводорода (в НТД мл/мин); m - скорость образования непредельных углеводородов в опытах со спиртом, либо скорость образования водорода при дегидрогенизации спирта или углеводорода (в мл/мин); Z2 и Z3 - относительные адсорбционные коэффициенты продуктов реакции (т. е. отношение адсорбционного коэффициента продукта реакции к адсорбционному коэффициенту исходного вещества); v - стехиометрический коэффициент: для спиртов v = 1, для тетралина v = 2. Z2 и Z3 рассчитывали кинетическим методом по формуле (3), вытекающей из общего кинетического уравнения Баландина:
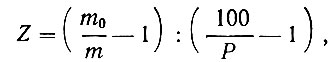
где m - объем водорода, выделившегося в единицу времени, или непредельных углеводородов в опытах с бинарной смесью (спирт - продукт реакции или углеводород - продукт реакции); m0 - то же, но в опытах с чистым веществом; Р - молярный процент спирта или углеводорода в бинарной смеси.
Этот метод определения относительных адсорбционных коэффициентов основан на том, что скорость реакции уменьшается, если к исходному веществу прибавить продукт реакции или постороннее вещество, так как последнее непроизводительно занимает часть каталитической поверхности.
Относительные адсорбционные коэффициенты Z на всех катализаторах определяли при 350-410° С для изопропилового спирта и при 480-530° С - для тетралина.
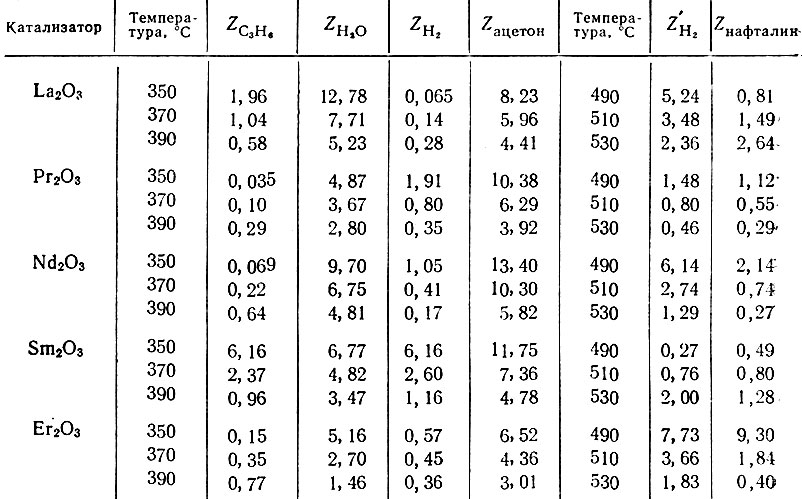
Таблица 3. Относительные адсорбционные коэффициенты Z продуктов реакции изопропилового спирта и тетралина
В табл. 3 приведены значения Z на всех изученных катализаторах при 350, 370 и 390° С. Относительные адсорбционные коэффициенты продуктов реакций преимущественно уменьшаются с увеличением температуры. Относительные адсорбционные коэффициенты воды ZH2J, ацетона Zацетон и водорода ZH2 в случае тетралина больше единицы, т. е. эти продукты вытесняют исходное вещество с каталитической поверхности и тормозят протекание соответствующих реакций тем больше, чем больше их Z. Относительные адсорбционные коэффициенты пропилена ZC3H6 водорода Z в случае спирта чаще всего меньше единицы, т. е. пропилен и водород адсорбируются слабее изопропилового спирта.
Если рассмотреть изменение, например, но на La2O3 №3, Рr2O3, Nd2O3, Sm2O3 и Еr2O3 при разных температурах, то из рис. 7 и табл. 3 видно, что величина Z на Рr2O3 и Sm2O3 меньше, чем на Lа2O3 и Еr2O3. Наиболее низки значения ZH2O на Еr2O3 при 370 и 390° С. Аналогичный график получается для изменения величины Zацетон на этих катализаторах при каждой из указанных температур. Интересно отметить, что кривые рис. 7 антибатны кривым рис. 5, показывающим изменение избирательности действия катализаторов в реакции разложения спиртов в зависимости от порядкового номера элемента, входящего в состав окисла. Так, Рr2O3 и Sm2O3 характеризуются более дегидрирующими свойствами, чем La2O3, Nd2O3 и, действительно, величины ZH2O и Zацетон на Рr2О3 и Sm2O3 меньше, чем на. La2O3 и Nd2O3, т. е увеличение относительных адсорбционных коэффициентов ацетона и воды способствует реакции дегидратации и уменьшает дегидрогенизацию. На Еr2O3 величины ZH2O и Zацетон наиболее низки при 390° С и, действительно, при этой температуре на Еr2O3 спирт дегидрируется в большей степени, чем на окислах Рr2O3 и Nd2O3. Таким образом, оказывается, что избирательность действия катализаторов зависит от величин Z продуктов реакции. Величины ZH2 и ZC3H6 невелики, например, при 370 и 390° С меньше единицы; такие коэффициенты мало влияют на избирательность ,и активность катализатора.
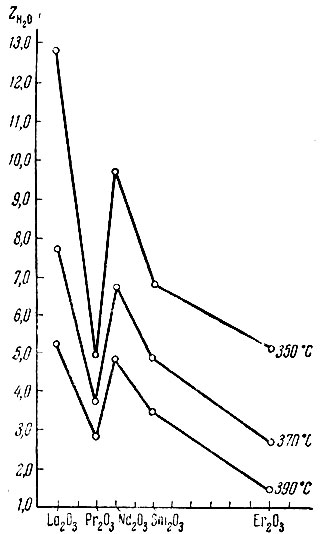
Рис. 7. Зависимость относительных адсорбционных коэффициентов воды от температуры на La2O3, Pr2O3, Nd2O3, Sm2O3 и Er2O3
Зная Z продуктов реакции, можно рассчитать по уравнению (2) истинные константы скорости, а по ним - истинные энергии активации реакции дегидрогенизации и EH2ист дегидратации EH2Oист изопропилового спирта и дегидрогенизации тетралина E'H2ист. Наблюдаемые энергии активации были рассчитаны, как и величины Z, из одной серии опытов при одной и той же активности катализатора и в одинаковом температурном интервале. Из табл. 4 видно, что величины истинных энергий активации Eист мало отличаются от наблюдаемых энергий активации, не более чем на 2,5 ккал/моль. В случае реакции дегидратации на Nd2O3 эта разница выше. Обычно Eист ≥ Eкаж
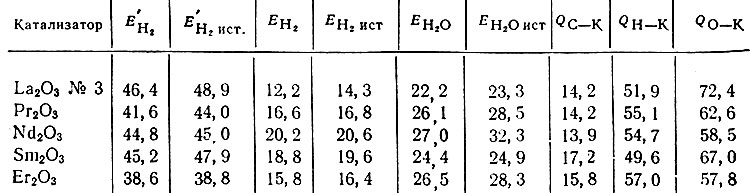
Таблица 4. Истинные энергии активации дегидрогенизации EH2ист и дегидратации ЕH2Oист изопропилового спирта и дегидрогенизации тетралина Е'H2ист. Наблюденные энергии активации тех же реакций ЕH2, ЕH2O, Е'H2. Энергии связей QC-К, QH-K и QO-K рассчитанные по Eист
Таким образом, определение энергии активации из температурной зависимости скорости образования газообразных продуктов в случае малых степеней превращений обычно дает близкую к истинной величину энергии активации.
Термодинамические функции адсорбционного вытеснения на каталитически активной поверхности
Относительные адсорбционные коэффициенты Z являются константами равновесия процесса адсорбционного вытеснения спирта или углеводорода продуктом его превращения, например водородом, или посторонним веществом с каталитически активной поверхности катализаторов. Поэтому по обычным термодинамическим формулам можно рассчитать разности величин свободной энергии ΔF, энтропии ΔS и энтальпии ДН этого процесса:
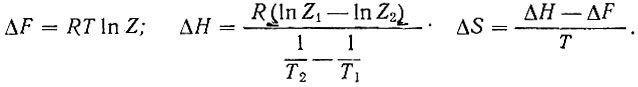 (4)
(4)Из табл. 5 видно, что во всех случаях разности теплот адсорбции Δλ(- ΔН) > 0, т. е. теплота адсорбции соответствующего продукта реакции больше теплоты адсорбции изопропилового спирта при дегидрогенизации и дегидратации и тетралина при его дегидрогенизации.
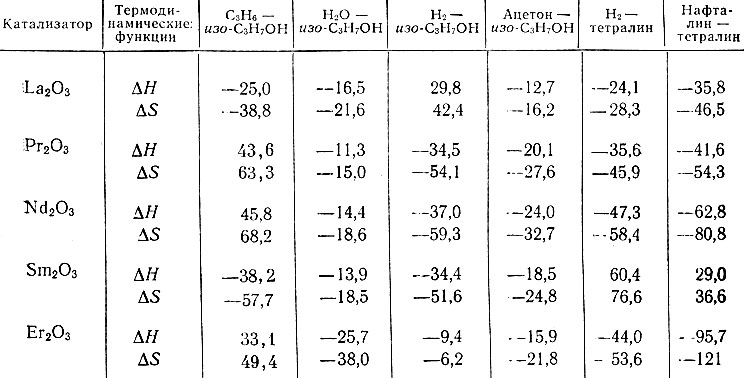
Таблица 5. Термодинамические функции ΔS и ΔН адсорбционного вытеснения изопропилового спирта и тетралина продуктами реакции
Разности энтропий адсорбции изученных веществ не зависят от температуры. Это означает, что при адсорбции молекулы теряют одни и те же степени свободы. Если проследить изменение ΔН и ΔS для всех продуктов реакции изопропилового спирта и тетралина на всех изученных катализаторах, то видно, что имеет место параллелизм между изменениями ΔН и ΔS во всех без исключения случаях. Существование параллелизма между ΔН и ΔS уже отмечалось ранее в случае превращения ряда спиртов на одном и том же катализаторе [19]. Наблюдается параллелизм при изменении ΔH и ΔS для пяти различных катализа- торов.
Относительные адсорбционные коэффициенты характеризуют процессы адсорбционного вытеснения на каталитически активной поверхности. На графике зависимости в координатах lg Z0 от ΔH точки хорошо укладываются на прямую, как в случае изопропилового спирта (рис. 6), так и тетралина. Из этого следует, что величины коэффициента распределения активных центров адсорбции спирта и тетралина на изученных катализаторах близки и функция распределения является экспоненциальной с близкими параметрами. Таким образом, наблюдается логарифмическая зависимость для величин lg Z0 и ΔH, аналогичная зависимости между lg К0 и Е. Необходимо отметить, что все точки как для той, так и для другой зависимости укладываются на одну и ту же прямую. Это свидетельствует о том, что распределение адсорбционных и каталитически активных центров одинаково, т. е. адсорбция и катализ протекают на одних и тех же активных центрах.
В работах Миначева, Маркова и других [20-23] по превращению пяти- и шестичленных цикленов на окислах Еr и Nd и дегидрогенизации циклогексана на окислах Nd, Sm, Gd, Но, Er, Tu, Yb, Y, а также в работе американских авторов [24] по дегидрогенизации циклогексана на окисях цериевой группы не наблюдалось зависимости каталитических свойств от магнитной восприимчивости указанных окисей. Это не противоречит нашим данным. В настоящей работе энергия активации дегидрогенизации тетралина также не зависит от магнитной восприимчивости рзэ, а изменяется симбатно ионному радиусу (см. стр. 116).
Литература
- А. А. Баландин. Изв. АН СССР, ОХН, № 4, 624 (1955).
- А. А. Баландин. ЖФХ, 31, 745 (1957).
- А. А. Баландин, А. А. Толстопятова. ЖФХ, 30, 1367, 1636 (1956).
- А. А. Толстопятова, А. А. Баландин. Проблемы кинетики и катализа, 10,. 351 (1960).
- А. А. Баландин, В. А. Ферапонтов, А. А. Толстопятова. Изв. АН СССР, ОХН, № 10, 1751 (1960).
- А. А. Толстопятова, А. А. Баландин. ЖФХ, 32, 1831 (1958).
- А. А. Толстопятова, А. А. Баландин. Сб. "Редкоземельные элементы". М., Изд-во АН СССР, 1958, стр. 307.
- А. А. Толстопятова, А. А. Баландин. ДАН СССР, 138, 1365 (1961).
- В. Н. Кондратьев. Усп. хим., 26, 861 (1957).
- Г. И. Леви, А. А. Баландин. Изв. АН СССР, ОХН, № 2, 157 (1960).
- В. В. Серебренников. Химия редкоземельных элементов, т. 1. Изд. Томск, ун-та, 1959, стр. 521. 1
- К. А. Гшнейдер. Проблемы современной металлургии, № 2 (50), 53 (1960).
- H. Landоll, R. Вornstеin. Zahlenwerte und Funktionen aus Physik, Chemie Astronomie, Geophysik und Technik. Bd. 1. Teil. 4. Berlin, Springer, 1955, S. 50, 114.
- А. А. Баландин. П. Тете ни. ДАН СССР, 132,577 (1960).
- М. Bou dart. J. Am. Chem. Soc., 74, 1531 (1952).
- D. A. Dоwden. J. Chem. Soc., 1950, 242.
- E. В. Maxted. J. Chem. Soc., 1949, 1987.
- J. Sheridan, W. D. Reid. J. Chem. Soc., 1952, 2962.
- О. К. Богданова, А. А. Баландин, А. П. Щеглова. Изв. АН СССР, ОХН, № 3, 425 (1961).
- X. М. Миначев, М. А. Марков, О. К. Щукина. Изв. АН СССР, ОХН, № 8 1507 (1961).
- X. М. Миначев, М. А Марков, О. К. Щукина. Изв. АН СССР, ОХН, № 9, 1665 (1961).
- X. М. Миначев, М. А. Марков, Г. А. Логинов. Нефтехимия, 1, 356 (1961).
- X. М. Миначев, М. А. Марков, О. К. Щукина. Нефтехимия, 1, 490 (1961)
- С. В. Me Cough, G. Houghton. J. Phys. Chem., 65, 1887 (1961).
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'