О роли комплексообразования при экстракционном выделении и разделении редкоземельных элементов и тория алкилфосфатами (З.А. Шека, Е.Е. Крисс, Э.И. Синявская)
Экстрагирование рзэ и тория осуществляется в настоящее время главным образом производными ортофосфорной кислоты.
Экстракция трибутилфосфатом
Из триалкилфосфатов для экстракции указанных элементов применяют трибутилфосфат (ТБФ).
Коэффициенты распределения рзэ при экстракции растворами ТБФ изменяются пропорционально третьей степени концентрации последнего, в связи с чем образующиеся при экстракции соединения представляются в виде сольватов состава Ln(NO3)3 × ЗТБФ [1-9]. При высоких концентрациях ТБФ наблюдается нарушение предельного кубического закона, что объясняют отклонением от идеальности, связанным с концентрациями веществ [5].
Пепцард с сотрудниками [7] высказали предположение, что состав экстрагируемого комплекса рзэ зависит от условий его образования и выражается формулой [Ln (ТБФ)a(Н2O)х-а](NO3)3, где а - функция концентрации HNO3; значение n: близко к 6 и зависит от порядкового номера элемента (для высоких - оно меньше 6, для низких - больше). Однако это предположение Пеппарда до сих пор не подтверждено. В более поздних работах отмечают, что соединения Ln(NO3)n × n ТБФ содержат постоянное число молекул комплексообразователя (ТБФ), не гидратированы и в неводной фазе нейтральны и не ионизированы [4, 10].
Торий экстрагируется ТБФ в виде комплекса Th(NO3)4 × 2ТБФ [1,4, 10-12].
Соединения Ln(NO3)3× 3ТБФ значительно менее устойчивы, чем соединения нитрата тория с ТБФ. Так, константы равновесия реакции
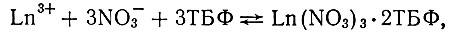
определенные для Се [8], La, Nd, Y и Yb [9], близки к единице; константа равновесия реакции
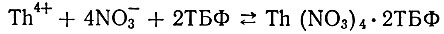
равна 36,0 ±2[11]. Этому соответствует и различие в коэффициентах распределения рзэ и тория. В случае невысокой кислотности растворов (до 3 мол/л), при которой определены указанные константы равновесия, коэффициенты распределения при экстракции 100%-ным ТБФ для рзэ
составляют 0,2-0,4 [6], для тория же он возрастает примерно до 80 [13]. С повышением содержания HN03 в растворах коэффициенты распределения рзэ и тория возрастают; при этом увеличивается также различие в экстрагируемости отдельных рзэ: коэффициенты распределения возрастают от лантана к лютецию. Так, при экстракции из 15 М HNO3, при прочих равных условиях, коэффициент распределения празеодима равен 0,81, гольмия - 94 [6], а тория-∼440 [13]. Одновременно с этим возрастает и коэффициент распределения HN03 [11, 13-20].
Состав комплексов нитратов рзэ с ТБФ при высокой концентрации HNO3 не установлен. Высказано предположение, что в этих условиях рзэ экстрагируются в виде не диссоциированных комплексных кислот состава H3Ln(NO3)6×xТБФ, подобно H2Ce(NO3)6 или Ln(NO3)3×xHNO3×TB0 [21, 22]. Напротив, Мак-Кей [4] отмечает, что для рзэ соединения, подобные H2M(NO3)6 или HMO2(NO3)3, не существуют и рзэ находятся в фазе ТБФ преимущественно в виде Ln(NO3)3 × 3ТБФ.
Изучение комплексообразования в системах Ln(NO3)3 - HNO3 - Н2O методами электромиграции, ионного обмена и распределения
между водной и неводной фазами показало, что ионы Ln и NO3 образуют комплексы состава Ln(NO3)3-nN, зависящие от концентрации нитрата рзэ и HNO3.
Методом электромиграции ионов на примере лантана показано, что при невысокой концентрации HNO3 в растворе существуют, главным образом, катионные формы. При повышении содержания HNO3 образуются анионные комплексы; при этом состав ионов зависит и от концентрации лантана. Так, при индикаторных количествах лантана катионные формы исчезают уже при концентрации HNO3 выше 3 мол/л. При содержании лантана 1-2 г-атом/л катионные формы существуют до концентрации HNO3 ∼ 7 мол/л.

Таблица 1. Электромиграция ионов La, Pr, Nd, Yb в 13М HNO3. Концентрация Ln(NO3)3 0,6 мол/л
Данные табл. 1 показывают, что с повышением атомного номера элемента перенос металла в анодную часть понижается и участие металла в переносе тока снижается. В 13 М растворе HNO3 при концентрации металла 0,6 г-атом/л лантан входит только в анионные комплексы, празеодим и неодим - в основном в анионные, a Yb, по-видимому,- преимущественно в состав нейтральных молекул.
Экспериментальные данные об ионном обмене на катионите КУ-2 и анионите ЭДЭ-10П позволили сделать вывод, что при индикаторных количествах рзэ в растворах, содержащих более 3 мол/л HNO3, образуются нитратные анионные комплексы рзэ, концентрация которых уменьшается с повышением атомного номера элемента. Параллельно с этим уменьшается и общая концентрация ионов в растворе, что свидетельствует, очевидно, о повышении устойчивости электронейтральных нитратных комплексов рзэ с увеличением их атомного номера.
Исследование поглощения рзэ катионитом и распределения рзэ между водным раствором HNO3 и неводной фазой (ТБФ и ССЦ) позволили определить константы устойчивости катионных и электро-нейтральных нитратных комплексов рзэ (табл. 2).

Таблица 2. Определение констант устойчивости комплексов LnNO2+3 Ln (NO3)3+ и Ln (NO3)3 различными методами
Несмотря на небольшое различие в константах устойчивости катионных нитратных комплексов рзэ, можно сделать вывод, что с увеличением их атомного номера устойчивость этих комплексов возрастает.
Полученные результаты позволяют сделать следующее заключение. Возникающие при высоком содержании HNO3 соединения HnLn(NO3)n+3 диссоциируют, образуя анионные комплексы. Диссоциация этих комплексов уменьшается с увеличением атомного номера элемента.
В этом же порядке повышаются коэффициенты распределения рзэ, что объясняется, по-видимому, ростом концентрации электронейтрального комплекса, который, сольватируясь ТБФ, образует экстрагирующееся соединение НnLn(NO3)n+3×3ТБФ. Кроме того, при высокой концентрации азотная кислота может служить и высаливателем, подобно нитратам, обычно применяемым при экстракции рзэ трибутилфосфатом.
Экстракция диалкилфосфорными кислотами
Для экстракции применяли растворы диалкилфосфорных кислот общей формулы (RO)2(O)OH: дибутил-, диамил-, ди-(2-этилгексил)- дифенилфосфорной кислоты [23-32], для тория - дибутил-, ди-(2-этил- гексил)-, ди-[n-(1,1,3,3 -тетраметилбугил) фенил]фосфорной кислоты [33, 34].
Взаимодействие между ионами металла и диалкилфосфорными кислотами (НА) при невысоком содержании HNO3, с учетом димеризации НА в неполярных растворителях, выражается схемой [28]:
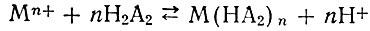 (1)
(1)или в случае рзэ, если принять во внимание диссоциацию димерных диалкилфосфорных кислот, как кислот одноосновных [29]:
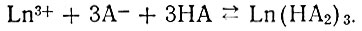 (2)
(2)Основанием для уравнений (1) и (2) послужила доказанная трехстепенная зависимость коэффициентов распределения рзэ от концентрации НА в неводной фазе.
Для тория найдено, что коэффициенты распределения находятся в двухстепенной зависимости от концентрации диалкилфосфорных кислот в неводной фазе и в четырехстепенной - от концентрации кислоты в водной фазе [33]. В связи с этим взаимодействие тория с диалкилфосфорными кислотами можно выразить схемой:
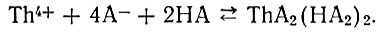 (3)
(3)При высоких концентрациях HNO3 в водном растворе возможна также реакция с образованием комплекса ThNO3(HA2)3 [33].
Нами определены константы равновесия К реакций (2) и (3) для Рг, Nd, Yb и Th при экстракции растворами дибутилфосфорной кислоты в ССl4 из водных азотнокислых растворов. Величины lg К соответственно равны 15,0; 15,4 ± 0,2; 18,6 ± 0,4; 26,3 ± 0,4. Дирссен [29] обнаружил, что для европия lg К равен 16,8.
С целью выяснения состава и условий образования комплексов рзэ с дибутилфосфорной кислотой приводим некоторые результаты исследований.
При синтезе дибутилфосфатов рзэ выделены соединения состава LnA3. По-видимэму, если в этом процессе и получаются сольватированные соединения типа Ln(HA2)3, то они разрушаются при промывании органическими растворителями.
Растворимость соединений LnA3 в растворах дибутилфосфорной кислоты в ССЦ на несколько порядков выше, чем в чистых четыреххлористом углероде, бензоле или этиловом спирте, и повышается с увеличением концентрации дибутилфосфорной кислоты. Очевидно, это результат дальнейшего взаимодействия LnA3 с дибутилфосфорной кислотой, которое может проходить по схеме
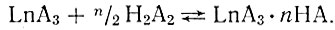
Экспериментальные данные показывают, что для иттербия n/2 = 1,5; это соответствует соединению Yb(HA2)3.
По мнению Пеппарда [271, соединения указанного состава как соль- ватированные, так и несольватированные,- внутрикомплексные.
Константа равновесия реакции (4), вычисленная нами по данным растворимости Yb(HA2)3 в растворах дибутилфосфорной кислоты в ССl4, равна 0,13 ± 0,04.
При дробной экстракции иттрия растворами дибутилфосфорной кислоты в ССl4 и молярном соотношении НА : Y, равном 3, достигается полное извлечение иттрия из водного раствора. Образующееся при этом соединение в неводной фазе растворяется мало; поэтому на границе фаз выделяется осадок не сольватированного соединения YA3. При экстракции, проводимой обычно в избытке дибутилфосфорной кислоты в неводной фазе, образуется соединение Y(HA2)3.
Комплексы рзэ и тория с дибутилфосфорной кислотой разрушаются кислотами с выделением мало растворимой, но устойчивой по отношению к ним дибутилфосфорной кислоты. Разложить дибутилфосфаты металлов можно нитрующей смесью или растворами щелочей. В последнем случае образуются гидроокиси рзэ и тория, а также растворимые в воде дибутилфосфаты щелочных металлов. Взаимодействие с щелочами используют для реэкстракции металлов из неводной фазы [35].
Снижение коэффициента распределения рзэ и тория при экстракции диалкилфосфорными кислотами с повышением концентрации HNO3 может быть объяснено разрушением экстрагируемого комплекса кислотой. Некоторое повышение коэффициентов распределения металлов при высокой концентрации кислот, по-видимому, связано с высаливающим действием HNO3, а также с образованием сольватов типа M(NO3)n×mHA, аналогично тому, как это предполагается для уранила [36].
Большое различие устойчивости диалкилфосфатов рзэ и тория может быть использовано для их экстракционного разделения.
В качестве примера приведем разделение рзэ и тория 0,15 М растворами дибутилфосфорной кислоты и ССl4 из сернокислых растворов при различной концентрации H2SO4. Каждый из рзэ извлекается при определенном содержании кислоты, которое с повышением атомного номера элемента следует увеличивать. Экстракцией смеси, содержащей 17% La, 19% Nd, по 16% Но и Еr и 32% Th (в сумме окислов), сначала из 6 N H2SO4, затем из 2 N H2SO4 получено три концентрата. Первый содержал 78% Th, второй - 69% Но и Ег, третий - 97% Nd и La.
Реэкстракцию проводили следующим образом. Рзэ цериевой группы осаждали из неводной фазы щавелевой кислотой, иттриевые элементы выделяли из растворов в дибутилфосфорной кислоте с NaOH. При этом достигали дополнительного разделения Nd и Но из смеси, содержащей 10 мол. % Ho(NO3)3 в сумме нитратов. После проведения одного цикла очистки (экстракции и реэкстракции) получено 99,4% Nd2O3 99,2%-ной чистоты и 98,7% Но2O3 99,8%-ной чистоты.
Таким образом, при экстракции дибутилфосфорной кислотой растворы тория и рзэ в H2SO4, полученные непосредственно после вскрытия монацита последней, могут быть использованы для группового разделения этих элементов.
Экстракция моноалкилфосфорными кислотами
Из моноалкилфосфорных кислот [(RO)Р(О) (ОН)2] Пеппардом с сотрудниками [37] с целью экстрагирования рзэ были изучены 2-этил- гексил- и n-(1, 1,3, 3 -тетраметилбутил)фенилфосфорные кислоты.
Коэффициенты распределения рзэ при экстракции моноалкилфосфорными кислотами, так же как при экстракции ди- и триэфирами, возрастают с увеличением атомного номера элемента. С повышением кислотности растворов коэффициенты распределения падают. Показано, что влияние указанных радикалов на коэффициенты распределения рзэ невелико. Коэффициенты распределения при экстракции моноэфирами выше, чем диэфирами, что особенно заметно при экстракции рзэ цериевой подгруппы.
По данным экстракции прометия установлено, что зависимость коэффициентов распределения от концентрации моно-(2-этилгексил) - фосфорной кислоты в толуоле является функцией первого порядка. В связи с этим экстрагируемому соединению предположительно приписывается состав Pm {Н[Н (RO) РO3]2}3.
Литература
- Т. V. Heal у, Н. А. С. МсК а у. Rec. trav. chim., 75, 730 (1956).
- Е. Gluckauf. Inorg. chim. Beige, 23, 1215 (1958).
- X. A. Mак - Кeй. Химия ядерного горючего. Доклады иностранных ученых на Международной конференции по мирному использованию атомной энергии. М., Гос- химиздат, 1956, стр. 498.
- Е. Неsfоrd, Н. А. Мс Кау. Trans. Farad. Soc., Б4, 573 (1958).
- D. Sea r gill, К. Alсосk, J. M. Fletcher, E. Hesford, H. A. C. McKay. J. Inorg. Nucl. Chem., 4, 304 (1957).
- K. Hesford, E. E. Jackson, H. A. C. McK a y. J. Inorg. Nucl. Chem., 9, 279 (1959).
- D. F. Peppard, W. J. Driscoll, R. I. Sironen, S. McCarty. J. Inorg. Nucl. Chem., 4, 326 (1957).
- В. В. Фомин, A. E. Коpтушeва, Т. И. Руденко. ЖНХ, 3, 2117 (1958).
- З. А. Шека, E. E. Крисс. ЖНХ, 6, 1930 (1961).
- G. Сarlesоn. Svensk kem. tidskr., 70, 55 (1958).
- В. В. Фомин, E. П. Mайоpова. ЖНХ, 1, 1703 (1956).
- E. Hesfоrd, H. A. C. Mc Kay, D. Sсargill. J. Inorg. Nucl. Chem., 4, 321 (1957).
- D. F. Peppard, G. W. Mason, J. L. Maler. J. Inorg. Nucl. Chem., 3, 215 (1956).
- К. Alсосk, S. S. Grimley, Т. V. Healy, J. Кennedy, H. A. C. McKay. Trans. Farad. Soc., 52, 39; 63, 633, (1956).
- А. С. Соловкин. ЖНХ, 2, 611 (1957).
- D. G. Tuсk. J. Chem. Soc., 19)58, 2783.
- В. Б. Шевченко, H. С. Повицкий, А. С. Соловки н, И. В. Шилин, К. П. Луничкина, 3. Н. Целткова. ЖНХ, 3, 2109 (1958).
- З. А. Шека, Е. Е. Крисс. ЖНХ, 4, 2505 (1959).
- Е. Hesford, Н. А. С. МсК а у. J. Inorg. Nucl. Chem., 13, 156 (1960).
- Т. Ishimori, К. Watanabe. Bull. Chem. Soc. Japan, 33, 1443 (1960).
- В. В. Фомин. Химия экстракционных процессов. М., Атомиздат, 1960, стр. 109.
- А. Н. Зеликман. Металлургия редкоземельных металлов, тория и урана. М., гостехиздат, 1960, стр. 164.
- R. Е. Соnniсk, S. W. Mayer. J. Am. Chem. Soc., 73. 1176 (1951).
- J. Hindman. Цит. no Trans. Farad. Soc., 54, 573 (1958).
- М. Г. Панова, H. E. Брежнева, В. И. Лeвин. Радиохимия, 2, 208 (1960).
- Е. М. Sсaddеn, R. Е. Bаllоu. Anal. Chem., 11, 1602 (1953).
- D. P. Рерраrd, G. W. Мasоn, W. J. Drisсоll, R. I. Sirоnеn. J. Inorg. Nucl. Chem., 7, 276 (1958).
- D. Dyrssen. Acta Chem. Scand., 11, 1277 (1957).
- D. Dyrssen, D. H. Liem. Acta Chem. Scand., 14, 1100 (1960).
- D. F. Peppard, G. W. Mason, J. L. Maier, W. J. Driscoll. J. Inorg. Nucl. Chem., 4, 334 (1957).
- З. А. Шека, E. E. Крисс. ЖНХ, 5, 2819 (1960); 6, 1930 (1961).
- E. H. Петрушевa, H. E. Брежнева, Г. В. Корпусов. Радиохимия, 2, 541 (1960).
- D. F. Peppard, G. W. Мasоn, S. Мс Саrty. J. Inorg. Nucl. Chem., 13, 138 (1960).
- Э. К. Хайд. Химия ядерного горючего. М., Гостехтеоретиздат, 1956, стр. 393.
- D. С. Мadigan. J. appl. Chem., 9, 252 (1959).
- Т. V. Healy, J. Kennedy. J. Inorg. Nucl. Chem., 10, 128 (1959).
- D. F Peppard, G. W. Mason, R. J. Sirоnen. J. Inorg. Nucl. Chem.. 10, 117 (1959).
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'