Основные итоги и перспективы работ в области хроматографического разделения смесей редкоземельных элементов (М.М. Сенявин)
Первая работа о хроматографическом разделении смесей рзэ была опубликована около 25 лет назад [1]. За прошедшее время в этом направлении было проведено столь много исследований, что невозможно сделать более или менее полный их обзор. Поэтому ограничимся лишь наиболее важными вопросами.
Для удобства все исследования в области хроматографического разделения смесей рзэ можно разделить на три периода.
С 1936 по 1943 г. (отчасти продолженный работами Линднера, 1946 г.) -период неудачных попыток разделения смесей рзэ способом простого вытеснения - промывания колонки растворами минеральных кислот или солей.
С 1943 г.- период отыскания принципа хроматографического разделения смесей рзэ - использования комплексообразующих реагентов и усовершенствования этого способа (в настоящее время - преимущественно на аналогах рзэ - трансурановых элементах).
С 1944 г.- период перенесения хроматографического способа разделения смесей осколочных рзэ на весомые количества. Развитие и усовершенствование этого направления интенсивно продолжается.
Не следует думать, что расширение масштабов хроматографического процесса, перенесение методик с невесомых "осколков" на лабораторные и, тем более, заводские масштабы - дело, автоматически решающееся и простое. Каждая из этих задач характеризуется своими требованиями. В случае осколков необходимо крайне быстро (в часы или минуты) количественно разделить изучаемую смесь рзэ. Цель же препаративных задач, особенно крупного масштаба,- экономично, по сравнению с другими методами, разделить смесь, содержащую нередко все рзэ, и получить при этом продукты, удовлетворяющие техническим условиям по чистоте и прейскуранту по стоимости.
В гидрометаллургическом процессе рзэ находятся в виде растворов солей, представляющих собой сильные, высоко диссоциированные электролиты. Поэтому, естественно, что в качестве сорбента для разделения их смесей употребляются преимущественно ионообменные смолы.
При разделении смесей рзэ чаще всего используют катиониты, что связано с катионной природой растворов солей этих элементов. Кроме того, именно в случае катионитов относительно слабее проявляется явление вытеснения, смазывающее картину разделения за счет различий в устойчивости комплексных соединений. Это объясняется тем, что трехзарядные катионы рзэ очень слабо вытесняются однозарядными ионами аммония или водорода. В то же время в случае разделения на анионитах, например, однозарядные анионы комплексов рзэ с ЭДТА легко вытесняются двух или трехзарядными ионами ЭДТА из промывающего раствора, что снижает степень хроматографического разделения. Тем не менее, разделению смесей рзэ на анионитах уделяется недостаточно внимания. Вследствие общеизвестного обращения порядка вымывания рзэ с анионитов они могли бы оказаться полезными при доочистке, например, элементов с нечетными порядковыми номерами. Если учесть дополнительно, что рзэ выходят из колонки катионита в комплексной анионной форме, то задача использования анионитов в общем цикле хроматографического разделения смесей рзэ значительно упрощается. Конечно, как указывалось выше, на анионитах можно работать лишь с разбавленными растворами комплексообразующих реагентов для уменьшения эффекта вытеснения.
Для целей хроматографического разделения смесей рзэ были опробованы самые различные катиониты. Подбор катионита,- по-видимому, первый этап любого исследования в области хроматографического разделения смесей рзэ. Некоторые из описанных в литературе катионитов, использующихся при хроматографическом разделении смесей рзэ,. приведены ниже.

В основном работы проводятся на монофункциональных сополиме- ризациоиных катионитах типа КУ-2. Детальное же исследование возможности разделения смесей рзэ на катионите НСК, по-видимому, не вполне оправдано [2]. Применение сильноосновных катионитов не случайно, поскольку процесс нередко проводят в кислых средах, как необходимых для предотвращения гидролиза рзэ, так и возникающих, например, при разделении смесей рзэ с комплексонами. Конечно, монофункциональный сульфокатионит типа КУ-2 - это катионит практически неселективный, но из-за предельной близости свойств рзэ трудно рассчитывать на создание катионита селективного действия. То обстоятельство, что селективность катионита типа КУ-2 практически равна нулю, иллюстрируется литературными данными по коэффициентам разделения, которые равны практически единице для любой пары: соседних рзэ:
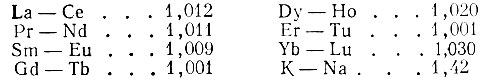
Сказанное, однако, не означает, что не могут быть созданы катиониты, селективные для рзэ. Возможно, селективными могли бы оказаться катиониты, содержащие несколько остатков аминоацетата типа НТА или ЭДТА. Промывающим раствором в этом случае мог бы служить не дорогой и в известной степени не полностью регенерируемый комплексон, а, например, соляная кислота или какой-либо буферный раствор. Описаны иониты такого типа [3]. Можно было бы пожелать синтетикам усилить и ускорить работы по созданию хелатных ионообменных сорбентов, а также увеличить емкость сульфокатио-нитов. Это пожелание не означает, конечно, что емкость существующих сульфокатионитов очень мала. Она равна примерно 5 мг-экв/г (3 мг-экв/мл), что соответствует примерно 1 М раствору рзэ. Это практически отвечает или близко к полному насыщению емкости такого эффективного экстрагента, как ТБФ (считая состав комплекса рзэ o 3 ТБФ), широко используемого в экстракционных процессах, отличающихся высокими концентрациями в каждой из фаз. Тем не менее, дальнейшее повышение емкости сульфокатионита весьма желательно, ибо позволило бы увеличить удельную загрузку смолы или уменьшить ее количество и габариты аппаратуры. По-видимому, возможный путь повышения емкости сульфосмол - это не столько увеличение степени сульфирования*, сколько облегчение органического каркаса смолы, например использование полиэтилена вместо стирола. Последнее могло бы повысить емкость сульфокатионита в 2-3 раза.
* (При расчете катионита, содержащего одну сульфогруппу на каждое бензольное кольцо, получается емкость около 5,2 мг-экв/г, ввести же две сульфогруппы в одно и то же кольцо невозможно вследствие потери смолой химической устойчивости.)
Наряду с дальнейшим улучшением качества ионообменных смол настало время подойти к рассмотрению возможности разделения смесей рзэ в препаративном плане с комплексообразующими реагентами, но без ионообменных сорбентов. В этом отношении имеются очень интересные работы Шведова с сотрудниками [4, 5] по разделению радиохимических количеств рзэ в электрическом поле на инертном носителе, например песке. Авторам удалось методом электрофореза практически полно и быстро разделить некоторые соседние рзэ. Из изученных комплексообразующих реагентов наилучшие результаты были получены с ЭДТА. Не исключено, что и в этом случае окажется трудным, но возможным переход от разделения радиохимических и осколочных количеств к количествам, представляющим препаративный интерес.
Наряду с сорбентом, вторым компонентом хроматографической системы является промывающий раствор, его природа, концентрация и величина pH. Необходимо подчеркнуть, что при хроматографическом разделении смесей рзэ указанный параметр - наиважнейший. Это связано с безуспешными попытками разделения смесей рзэ способом простого вытеснения, возможностью быстрого количественного вымывания с катионита трехзарядных ионов 0,01 N раствором лимонной кислоты (для простого вытеснения трехзарядного катиона требуется по меньшей мере 2-3 N раствор), наконец, с крайне резкой зависимостью степени хроматографического разделения смесей рзэ от pH (например, в случае использования лимонной кислоты). Роль процесса комплексообразования видна и из неудачных опытов по хроматографическому разделению смесей рзэ на сильносшитых смолах, в которых селективность по другим ионам, особенно щелочных металлов, значительно выше.
В случае рзэ выбор промывающего раствора имеет особое значение, и можно утверждать, что степень разделения их смесей определяется в идеальном случае соотношением констант неустойчивости соответствующих комплексов. Конечно, при неправильном выборе условий разделение не достигается даже с самым лучшим комплексообразующим реагентом. Степень разделения, равная отношению констант неустойчивости, это предел возможности и мечтаний экспериментатора.
Однако, к сожалению, выбор комплексообразующих реагентов совершается эмпирически. Теория, которая позволила бы предсказать наилучший комплексообразующий реагент, отсутствует. Не менее трудно выбрать условия применения данного комплексообразующего реагента его концентрацию и pH раствора. В основном положение очень изменилось с 1947 г., когда американские исследователи утверждали, что в области хроматографии рзэ "получение хороших результатов - часто не наука,а мастерство" [6].
В настоящее время как будто бы надежно можно предсказать лишь одно требование к комплексообразующим реагентам: они должны давать устойчивые комплексы с компонентами разделяемой смеси. В противном случае явление вытеснения значительно смазывает эффект разделения. О значении эффекта вытеснения наглядно свидетельствует работа по разделению смесей Rb и Cs 0,5-1%-ной ЭДТА [7], так как точно известно, что эти элементы с ЭДТА не образуют комплексов.
Как было показано и объяснено систематическими исследованиями Рябчикова и Терентьевой [8, 9], лимонная кислота - первый из комплексообразующих реагентов, использованных при хроматографическом разделении смесей рзэ,- образует комплексы очень высокой устойчивости. Однако дальнейшие поиски комплексообразующих реагентов не всегда следовали правилу повышения устойчивости. Некоторые комплексообразующие реагенты, использующиеся при хроматографическом разделении смесей рзэ, приведены на стр. 259. Так, для разделения осколков вместо лимонной кислоты стали применять одноосновные а-оксикислоты, образующие менее прочные комплексы.
В области препаративной, особенно в первое времяу применяли самые различные комплексообразующие реагенты. В настоящее время наметились два способа использования комилексообразующих реагентов: без замедлителей и с замедлителями (рис. 1).
В опытах без замедлителей наиболее широко применяли, а отчасти и применяют до сих пор лимонную кислоту. К сожалению, теория процесса совершенно неизвестна. До сих пор отсутствуют надежные данные о константах устойчивости различных цитратных комплексов рзэ, что во многом объясняется многоосновностью кислоты, и соответственно, сосуществованием большого числа комплексных форм.
Условия применения лимонной кислоты также выбирали эмпирически. Как известно, все первоначальные опыты проводили с 5%-ной лимонной кислотой при pH ∼ 3 [10]; концентрация рзэ в растворе была равна примерно 1 г/л. Важно отметить, что при этом расходуется колоссальный избыток лимонной кислоты, ибо, как показывает расчет, в процесс комплексообразования входит не более 5-10% от введенного ее количества*.
* (5%-ный раствор лимонной кислоты в предположении, что состав комплекса соответствует формуле LnCit3-2, эквивалентен 0,12М раствору рзэ (∼18 г рзэ/л.).)
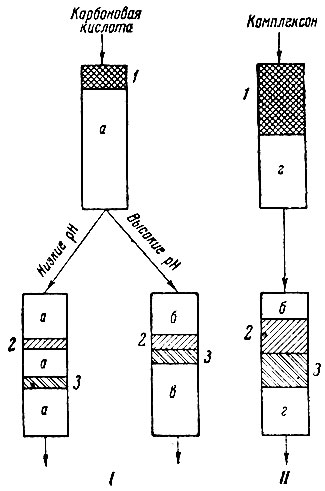
Рис. 1. Схемы процессов хромато-графического разделения смесей рзэ: I - без замедлителей; II - с замедлителями. 1 - ΣLnl и Ln2; 2- Lnl; 3 - Ln2; а - катионит в смешанной Н - NH4-форме; б- катионит в NH4- форме; в - катионит в Н-форме; г - катионит в Сu - форме
В то же время, несмотря на столь малое использование кислоты при этих значениях pH, в процессе комплексообразования участвуют не только цитрат-ионы, но и ионы HCit2-, а частично даже H2Cit-. Последнему ранее придавали особое значение, говорили лишь о высокой избирательности системы с ионами H2Cit-, да и комплексам приписывали формулу Me(H2Cit)3 [11].
В целях повышения степени использования лимонной кислоты Спеддинг [12, 13] предложил проводить опыты при более высоких pH (относительное увеличение содержания цитрат-ионов), что компенсировали понижением концентрации кислоты:

Использование разбавленных растворов в технологии оказалось не совсем целесообразным, ибо получали малые концентрации рзэ в фильтратах, что усложняло и удорожало их переработку (большие объемы, расход осадителя, потери). Однако в результате этих исследований Спеддингу [14] удалось открыть новый способ способ замедлителей. При высоких pH, когда присутствует и образует комплекс лишь цитрат-ион, водород смолы тормозит, замедляет перемещение рзэ вследствие разрушения комплекса. Зона рзэ движется за зоной ионов водорода, выстраиваясь в этом процессе в соответствии с константами не-устойчивости. Метод замедлителей оказался технологически очень перспективным, ибо позволил резко расширить круг используемых комплексообразующих реагентов, причем при сравнительно высоких значениях pH. Это позволило повысить и концентрации рзэ в фильтратах в среднем до 10 г/л. В качестве комплексообразующего реагента было рекомендовано использовать соли полидентатных аминополиуксусных кислот, предложенных в 1951 - 1952 гг. Фитчем и Расселом [15], а также Виккери [16], но забракованных вследствие нерастворимости соответствующих свободных кислот. В качестве замедлителей первоначально предлагали ионы железа и меди [17]. Впоследствии был резко расширен круг замедлителей и число комплексообразующих реагентов. Оказалось, что в качестве замедлителей можно использовать все элементы середины четвертого периода от Мn до Zn, образующие прочные аммиакаты, а в качестве комплексообразующих реагентов - многообразные комплексоны. Некоторые из них были указаны выше.
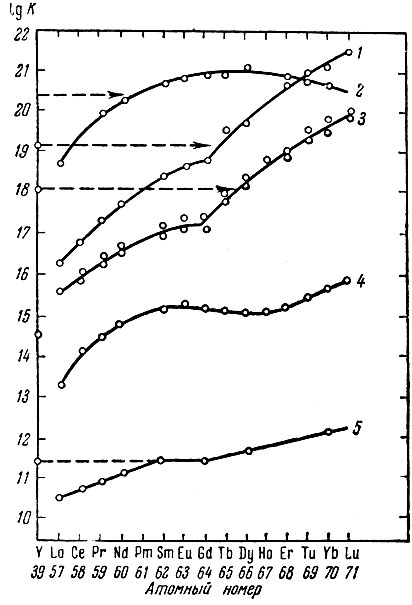
Рис. 2. Константы устойчивости комплексонов, использующихся при хроматографическом разделении смесей рзэ.1 - циклогександиаминтетраацетат; 2 - диэтилентриаминпентаацетат; 3 - ЭДТА; 4- оксиэтил-этилендиаминтриацетат; 5 - НТА
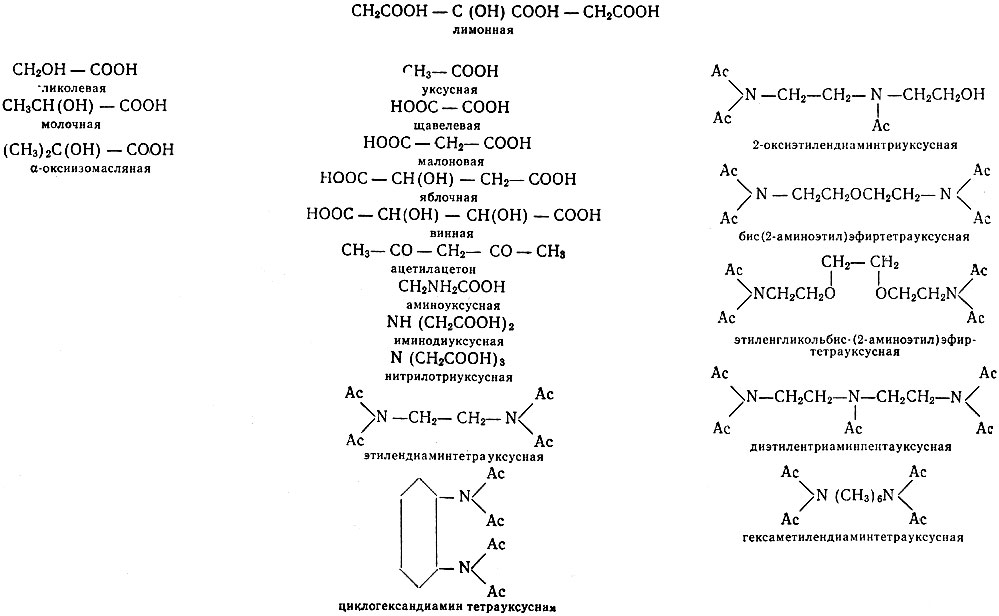
Органические кислоты, используемые в качестве комплексообразующих реагентов при хроматографическом разделении смесей рзэ
С какими же комплексонами были получены наилучшие реаультаты? Данные об устойчивости их комплексов с рзэ приведены на рис. 2. Как видно, наиболее прочные комплексы образуют циклогександиамин- тетраацетат и диэтил ентр и а м инпента а цетат. Но у последнего в области гадолиния, где всегда имеется излом, наблюдается даже максимум, что делает разделение ряда смесей при его помощи невозможным. Большие различия величин констант нестойкости рзэ наблюдаются у ЭДТА и ЦГДТА. Важное преимущество оксиэтилендиаминтриацетата - отсутствие высаливания, т. е. его сравнительно хорошая растворимость.
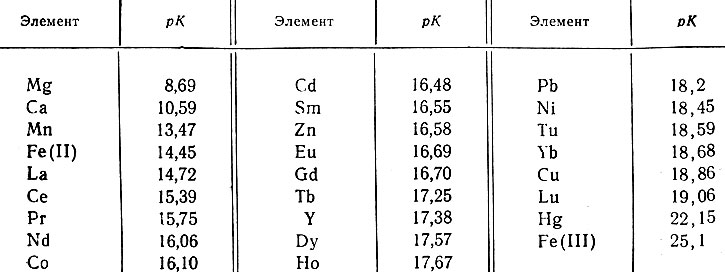
рК комплексов рзэ и ионов-замедлителей ЭДТА
Интересно отметить несколько различное положение иттрия в ряду рзэ при использовании разных комплексообразующих реагентов. Так, в случае лимонной кислоты иттрий располагается между диспрозием и гольмием, в случае ЭДТА - в районе тербия, в случае НТА - самария, а в случае диэтилентриамшшентаацетата даже в районе неодима. Это делает целесообразным ори разделении смесей иттриевой подгруппы последовательно использовать различные комплексообразующие реагенты.
На практике наиболее широко используют ЭДТА и НТА, возможно, ввиду сравнительной простоты их синтеза. В случае ЭДТА сравнительные данные о константах устойчивости рзэ и различных замедлителей приведены в таблице.
Таким образом, в настоящее время в результате эмпирических исследований имеются некоторые рекомендации по разделению смесей рзэ при помощи комплексонов и ионов-замедлителей. Найденные результаты, очевидно, не оптимальны. В частности, очень важно было бы определить условия, при которых концентрация рзэ в фильтратах доходила бы до 30-50 г/л. Следовало бы всемерно развивать теорию процесса хроматографического разделения смесей рзэ в целях предсказания оптимальных комплексообразующих реагентов и условий их использования. Это требует коллективных усилий химиков-неоргаников, физико-химиков и химиков-синтетиков.
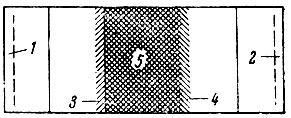
Рис. 3. Принципиальная схема электрохимического концентрирования рзэ в присутствии комплексообразующих реагентов. 1 - катод; 2 - анод; 3 - катионитовая мембрана; 4 - анионитовая мембрана; 5 - Ln + Ln2 (с большим порядковым номером)
Помимо равновесных характеристик, степень хроматографического разделения существенно зависит от кинетики процесса. Как известно, в случае простого вытеснения кинетика, как правило, имеет диффузионный характер. Кинетика процессов, связанных с комплексообразованием, может быть существенно более медленной. Неслучайно при разделении осколочных рзэ используют одноосновные оксикислоты - в этом случае процесс комплексообразования, возможно, проходит скорее. Известны литературные данные, свидетельствующие о сравнительно медленном взаимодействии рзэ с комдлекоонами [18]. Не исключено, что в ряде случаев кинетика при этом может иметь и химический характер. Следовало бы рекомендовать (постановку работ по детальному (в тождественных условиях) сравнительному изучению кинетики в отсутствие комплексообразующих реагентов и в присутствии различных оксикислот и комплексонов.
Кинетика процесса имеет существенное значение при выборе таких параметров опыта, как зернение сорбента и скорость течения раствора В последнем случае надо различать так называемые линейные и объемные скорости. Как было показано ранее [19], в случае процессов без комплексообразующих реагентов целесообразно проводить опыты при малых объемных скоростях, но при больших линейных скоростях. Это сводится практически к использованию длинных и узких колонн. К сожалению, эти параметры для случая разделения смесей рзэ пока не изучены. Колонны конструируют совершенно произвольно; при этом не учитывают в должной мере как чисто кинетические, так и гидродинамические параметры. Следовало бы детально рассмотреть вопрос об аппаратуре для хроматографических опытов.
Благодаря интенсивным исследованиям, даже несмотря на эмпиричность хроматографического разделения смесей рзэ, ионообменная хроматография из метода разделения осколков выросла в важный технологический метод, который в настоящее время незаменим для получения препаратов высокой чистоты. Более того, в случае процессов сравнительно невысокой производительности он может рассматриваться почти как единственный. Но при проведении крупномасштабных процессов, это, по-видимому, нецелесообразно: во-первых, широко используют концентраты рзэ; во-вторых, для повышения экономичности хроматографический метод следует сочетать с методами предварительного концентрирования (основное осаждение, кристаллизация, частичное комплексообразование, экстракция [20-24]). Для целей концентрирования, вероятно, перспективен и метод электродиализа [25] (схема изображена на рис. 3) частично закомплексованной смеси рзэ, характеризующийся высокой производительностью. Главное в такой работе это определение целесообразных условий сочетания методов предварительного концентрирования с методом ионообменной хроматографии, позволяющих наиболее полно выявить преимущества каждого из них.
Наряду с установлением такого рационального сочетания необходимо продолжать работу по усовершенствованию хроматографии и дальнейшему выявлению ее возможностей. Для этого, по-видимому, было бы необходимо развитие работ в следующих направлениях.
- Создание теории метода, превращение его из эмпирического в современный научный метод. Это требует всестороннего изучения статики, кинетики и динамики обмена смеси ионов; состава и устойчивости комплексов, используемых или перспективных для хроматографии; статики, кинетики и динамики смесей в присутствии комплексообразующих реагентов.
- Синтез ионообменных смол высокой емкости, лучшей кинетики, а также хелатных смол. Представляется необходимой организация выпуска опытных партий новых смол, чтобы можно было сравнивать их в полупромышленных условиях.
- Синтез и получение разнообразных комплексообразующих реагентов в количествах, достаточных для экономически обоснованного, полупромышленного выбора наилучших образцов.
- Разработка теории, конструкции, а также изготовление разнообразной хроматографической аппаратуры для правильного оформления процесса хроматографического разделения смесей рзэ.
- Разработка методов непрерывного определения концентрации отдельных рзэ в ходе хроматографического процесса.
Литература
- М. М. Сенявин. Сб.: "Ионный обмен и его применение". М., Изд-во АН СССР, 1959, стр. 127.
- Я. Я. Додонов, В. П. Храмов, В. С. Колосова. Сб.: "Редкоземельные элементы". М., Изд-во АН СССР, 1958, стр. 121.
- Е. Вlasius, G. Оlbrich. Z. anal. Chem., 152, 81 (1956).
- В. П. Шведов, А. В. Степанов. Радиохимия, 1, 162, 668 (1959).
- А. В. Степанов. Автореферат диссертации. Ленингр. гос. ун-т, 1960.
- Сб.: "Хроматографический метод разделения ионов". М, ИЛ, 1949.
- А. П. Смирнов - Аверин, Г. Б. Ко ста рев, Н. Н. Крот. ЖАХ, 12, 313 (1957).
- Д. И. Рябчиков, Е. А. Терентьева. ДАН СССР, 51, 287 (1946).
- Е. А. Терентьева. Диссертация. МГУ, 1951.
- Д. И. Рябчиков, Е. А. Терентьева. Усп. хим., 24, 260 (1955).
- Д. Шуберт. Сб.: "Ионный обмен". М., ИЛ, 1950.
- F. Sреdding, F. Fulmer. I. Butler, I. Powell. J. Am. Chem. Soc., 72, 2349 (1950).
- F. Spedding, F. Fulmer, I. Powell, I. Вuller. J. Am. Chem. Soc., 72, 2354 (1950).
- F. Spedding, I. Powell, E. Wheelright. J. Am. Chem. Soc., 76, 612 (1954).
- F. Fitch, D. Russel. Canad. J. Chem., 29, 363 (1951).
- R. Vickery. J. Chem. Soc., 1952, 1895.
- Сб.: "Редкоземельные металлы". М., ИЛ, 1959.
- Radioisotopes in scientific research, v. 2. N. Y., Pergamon Press, 1959, p. 326.
- М. М. Сенявин, E. П. Жиров. Зав. лаб., 25, 843 (1959).
- Д. Иост, Г. Рэссель, К. Гарнер. Редкоземельные элементы и их соединения. М., ИЛ, 1949.
- Р. В. Котляров, Г. П. Кожемяко. Сб.: "Редкоземельные элементы". М., Изд-во АН СССР, 1958, стр. 52, 62.
- Д. И. Рябчиков, H. С. Вагина. ЖНХ, 5, 356 (1969).
- Сб.: "Экстракция", № 1 и 2. М., Атомиздат, 1962.
- Г. В. Корпусов, Е. H. Патрушева и др. Наст, сб., стр. 224.
- Б. H. Ласкорин, H. М. Смирнова, М. И. Гаитман. Ионообменные мембраны и их применение. М., Атомиздат, 1961.
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'