2. Стационарная кинетика катализа гидрогеназами
В работе [327] изучена стационарная кинетика выделения молекулярного водорода из восстановленных органических соединений, обладающих достаточно отрицательным окислительно-восстановительным потенциалом, таких как метилвиологен, бензилвиологен, нейтральный красный.
Исследование кинетики образования водорода из этих соединений под действием гидрогеназы проводили в анаэробных условиях в избытке восстанавливающего агента - дитионита натрия. При этом некоторые стадии механизма катализа ферментом становятся необратимыми, что существенно упрощает формальнокинетический анализ механизма реакции. В отсутствие органических субстратов из дитионита натрия под действием гидрогеназы не наблюдается выделение водорода. Истинными субстратами гидрогеназы в системе дитионит натрия - органические субстраты - гидрогеназа являются восстановленные формы органических молекул. При концентрациях, при которых дитионит натрия находится в избытке по сравнению с субстратом, скорость реакции выделения водорода не зависит от концентрации дитионита. Дитионит натрия переводит субстраты в восстановленную форму и на начальных этапах реакции практически не расходуется.
Была исследована зависимость начальной скорости выделения водорода от концентрации субстрата при использовании в качестве субстратов метилвиологена и бензилвиологена. Зависимость скорости реакции от концентрации этих субстратов описывается уравнением типа уравнения Михаэлиса-Ментен. Экспериментальные данные достаточно хорошо спрямляются в координатах уравнения Лайнуивера - Берка. Из зависимостей такого характера найдены параметры υмакс и KМ (табл. 17).
![Таблица 17. Значение vмакс и КM в уравнении скорости реакции от концентрации восстановленных форм субстратов для гидрогеназы Th. roseopersicina [327]](pic/000074.jpg)
Таблица 17. Значение υмакс и КM в уравнении скорости реакции от концентрации восстановленных форм субстратов для гидрогеназы Th. roseopersicina [327]
Обнаружено, что гидрогеназа из Th. roseopersicina обладает достаточно широкой специфичностью. Некоторые органические соединения, субстраты гидрогеназы, обладают сравнительно отрицательным окислительно-восстановительным потенциалом; в этом случае можно наблюдать реакцию выделения водорода из восстановленных форм этих соединений. В частности, субстратом фермента является восстановленная форма индикатора нейтрального красного.
Зависимость начальной скорости выделения водорода при использовании в качестве субстрата нейтрального красного от его концентрации имеет сложный характер. При высоких концентрациях субстрата наблюдается уменьшение скорости реакции (ингибирование избытком субстрата). Значения υмакс и КМ для нейтрального красного, приведенные в табл. 10, вычислены с учетом эффекта ингибирования субстратом [327].
Из таблицы 17 видно, что в то время как значения КМ существенно различаются, величины υмакс, найденные при использовании равных концентраций фермента, практически совпадают. Это указывает на то, что величины каталитических констант скорости kкат для изученных субстратов равны между собой. Таким образом, лимитирующие константы скорости в катализе исследуемой гидрогеназы практически независимы от природы субстрата, и, следовательно, параметр υмакс включает константы (или константу) скорости и равновесия, не связанные со взаимодействием субстратов с активным центром фермента, а характеризующие общие для всех субстратов стадии (например, реакцию взаимодействия фермента с молекулярным водородом).
Важно отметить, что постоянство kкат при широкой вариации КМ характерно для многих двухсубстратных реакций. Это означает, что при "насыщении" активного центра фермента одним из субстратов лимитирующей стадией становится взаимодействие с другим субстратом. При этом величина υмакс(kкат) естественно не зависит от природы первого субстрата.
Нами исследованы зависимости скорости реакций, катализируемых гидрогеназами, от pH. Ионы водорода служат субстратами в реакции образования молекулярного водорода. Поэтому исследование этих зависимостей может дать принципиальную информацию о механизме процесса.
Зависимость υмакс и КМ для реакции образования водорода из восстановленной формы метилвиологена приведена на рис. 16. Скорость реакции падает при низких значениях pH вследствие увеличения константы Михаэлиса, а при высоких значениях pH - уменьшения параметра υмакс. Зависимость скорости реакции от концентрации ионов водорода контролируется ионогенными группами с рКа = 6,5 по КМ и рКа = 10 по υмакс [327].
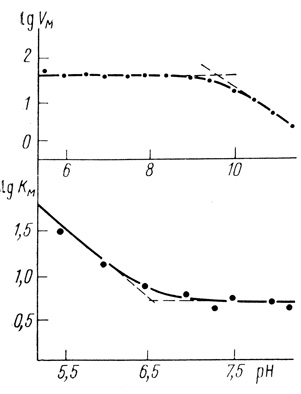
Рис. 16. pH-Зависимости максимальной скорости и константы Михаэлиса для реакции выделения водорода из восстановленной формы метилвиологена (25°С; 0,02 М ацетата натрия; pH 5-7,5; 0,02 М бората натрия (pH 7,5-12)
Зависимость Км от концентрации ионов водорода, по-видимому, связана с равновесием в системе восстановленная форма метилвиологена - ионы водорода. В работе [328] показано, что с рКа = 6,5 происходит присоединение протона к восстановленной форме метилвиологена. Известно, что протолитические равновесия, имеющие место с субстратом, проявляются в pH-зависимости константы Михаэлиса. Принципиальным для понимания механизма катализа гидрогеназами является анализ pH-зависимости максимальной скорости процесса.
На основе полученных экспериментальных данных возможно построение кинетических моделей действия фермента. Кинетическая модель, описывающая механизм действия гидрогеназы Th. roceopersicina, должна удовлетворять следующим экспериментальным фактам:
1) Зависимость скорости реакции от концентрации органических субстратов описывается уравнением типа уравнения Михаэлиса-Ментен.
2) Величины параметра kкат реакции выделения водорода практически не зависят от природы используемого органического субстрата при изменении свойств последнего в достаточно широком диапазоне.
3) В реакции выделения водорода протоны являются фактически "субстратами" гидрогеназы. Кинетическая модель должна описывать экспериментально полученную зависимость скорости реакции от концентрации ионов водорода.
При анализе механизмов действия ферментов важен вопрос о природе лимитирующей стадии процесса. Для выяснения природы лимитирующей стадии и доказательства участия в механизме реакции промежуточного соединения существует качественный подход, основанный на исследовании кинетики реакции с субстратами, существенно различающимися по структуре и реакционной способности [253, 254]. В том случае, если максимальные скорости реакции с участием таких субстратов близки или равны между собой, можно думать, что в механизме реакции для всех субстратов принимает участие одно промежуточное соединение, и именно реакция превращения этого интермедиата становится стадией, определяющей скорость.
Экспериментальные данные, полученные в настоящей работе, указывают на то, что аналогичная ситуация имеет место в механизме катализа гидрогеназой из Th. roseopersicina. Исследованные субстраты существенно различаются по структуре и термодинамическим параметрам в реакциях окисления-восстановления (см. табл. 17). Кинетические различия в поведении субстратов в неферментативных реакциях переноса электрона также очень велики. Например, восстановленная форма метилвиологена окисляется кислородом воздуха в миллисекундные интервалы времени [334], в то время как восстановленная форма нейтрального красного сравнительно устойчива, и процесс окисления ее на воздухе протекает в течение нескольких часов. Однако в ферментативной реакции выделения водорода при "насыщении" субстратом оба соединения обнаруживают приблизительно одинаковую реакционную способность. Это указывает на то, что механизм катализа гидрогеназой включает стадию образования интермедиата, общего для обоих субстратов, и при "насыщенных" концентрациях субстратов лимитирующей является реакция превращения этого интермедиата.
Основными предпосылками в построении и анализе кинетических схем каталитического действия гидрогеназы является достаточно обоснованное представление о гетеролитическом расщеплении молекул водорода под действием гидрогеназ [330-333], а также предположение о том, что протеолитические равновесия в системе устанавливаются сравнительно быстро.
Простейшая кинетическая модель. Представление о том, что механизм катализа гидрогеназами включает стадию гетеролитического расщепления молекулы водорода, следует в основном из опытов по кинетике изотопного обмена и орто-пара-конверсии водорода в присутствии гидрогеназ. Стадия гетеролитического расщепления молекулы водорода с участием фермента может быть записана в виде
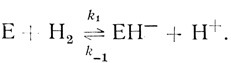 (2.22)
(2.22)Если стадию (2.22) принять за основу, то из стехиометрических соображений следует, что при реакции метилвиологена или бензилвиологена с водородом в присутствии гидрогеназы должны существовать две последовательные стадии взаимодействия промежуточного фермент-гидридного комплекса с органическими акцепторами (донорами) электрона. Метилвиологен и бензилвиологен, как это следует из полярографических данных [334], - одноэлектронные акцепторы. С учетом стадии депротонирования активного центра кинетическая схема действия гидрогеназы имеет следующий вид:
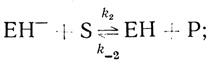 (2.23)
(2.23)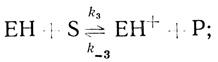 (2.24)
(2.24)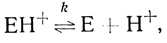 (2.25)
(2.25)где S и Р - окисленная и восстановленная формы субстрата соответственно; ЕН- - фермент-гидридный комплекс; ЕН и ЕН+ - промежуточные соединения в механизме катализа.
Механизм катализа (2.22-2.25) основан на следующих предпосылках: 1) активация молекулярного водорода гидрогеназами представляет собой стадию гетеролитического расщепления молекулы водорода с переносом двух электронов на активный центр фермента; 2) акцептирование электронов из активного центра органическими одноэлектронными субстратами представляет собой две последовательные стадии; 3) все стадии, входящие в механизм реакции, бимолекулярные; 4) стадия депротонизации активного центра (2.25) протекает в равновесном режиме. Данный механизм реакции является простейшим, который можно построить на основании изложенных выше предпосылок. Стационарная скорость выделения водорода в условиях избытка дитионита натрия, когда [S] = 0, дается уравнением типа Михаэлиса-Ментен, где
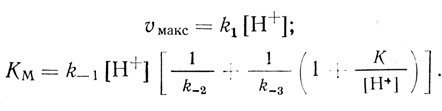 (2.26)
(2.26)Классификация механизмов. Описанный выше простейший механизм катализа гидрогеназой допускает ряд модификаций. Окислительно-восстановительные реакции протекают через элементарные стадии переноса электронов и протонов (или гидрид-атомов). В случае гидрогеназы в реакциях с участием одноэлектронных акцепторов (метилвиологен и бензилвиологен) электроны и протоны переносятся, очевидно, на различных стадиях, поскольку субстраты являются акцепторами (или донорами) только электрона. При этом важную роль играет последовательность стадий переноса протонов и электронов. Рассмотренная выше схема построена в предположении, что механизм реакции включает депротонирование частицы ЕН+. Однако депротонирование активного центра фермента при ионизации водорода может происходить с участием любой из промежуточных частиц - ЕН-, ЕН или ЕН+. Очевидно, что зависимость скорости реакции от концентрации компонентов и кинетических констант будет варьировать в зависимости от того, на какой стадии происходит депротонирование активного центра.
За основу при классификации возможных механизмов в работе [327] предложено использовать последовательность элементарных стадий переноса электронов и протонов. Для реакции восстановления органических соединений водородом после стадии образования фермент-гидридного комплекса реакция может протекать по трем механизмам: электрон-электрон-протон (еер), электрон-протон-электрон (ере) или протон-электрон-электрон (рее).
В таблице 18 приведены возможные модификации схем (2.23-2.25) и выражения для кинетических параметров kкат и КМ, соответствующие различным механизмам.
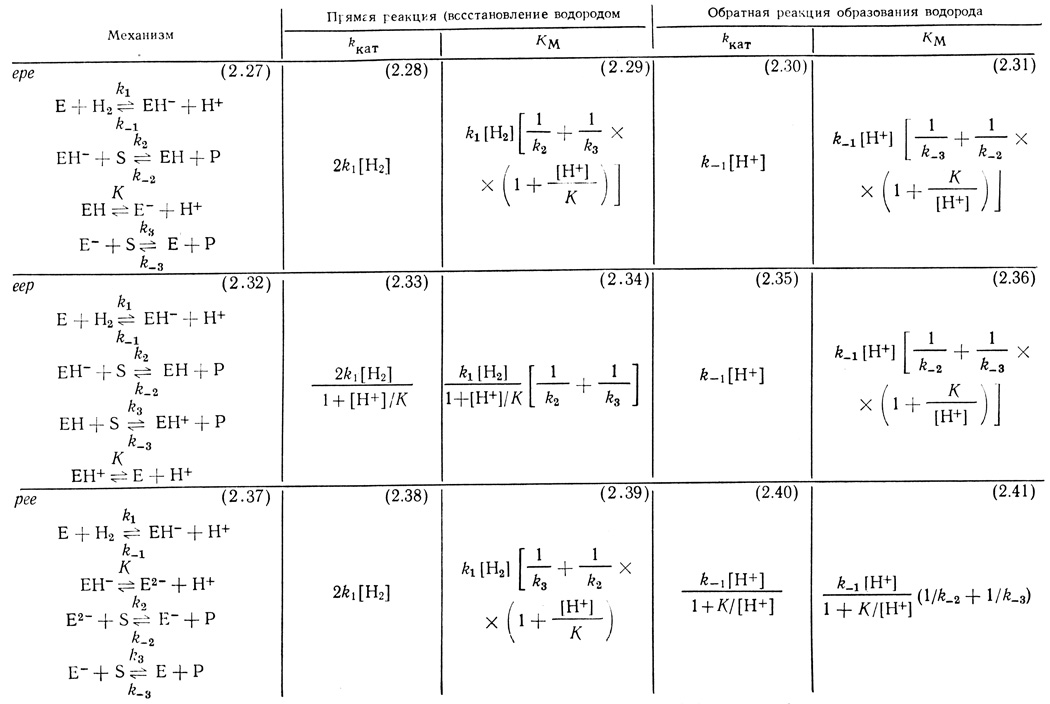
Таблица 18. Модификация схем (2,23-2,25) и кинетические параметры kкат и KМ реакции восстановления органических соединений водородом
Рассмотренная кинетическая модель достаточно хорошо описывает некоторые особенности катализа гидрогеназой. Во-первых, зависимости скорости реакций от концентраций органических субстратов имеют классический вид уравнения Михаэлиса. Во-вторых, параметры kкат и соответственно υмакс определяются константами скорости и равновесия, не включающими параметры взаимодействия активного центра с органическими субстратами. Из уравнений (2.30), (2.35) и (2.40) следует, что kкат в реакции выделения водорода определяется только константой скорости взаимодействия фермент-гидридного комплекса с протоном и не включает в себя константы скорости k-2 и k-3, которые связаны с природой субстрата.
Однако обсуждаемая кинетическая модель не описывает зависимости максимальной скорости реакции от концентрации ионов водорода. Действительно, из уравнений (2.30) и (2.35) следует, что υмакс должна линейно зависеть от концентрации ионов водорода, а для случая механизма рее линейная зависимость должна переходить в квадратичную при низких концентрациях ионов водорода. Это не соответствует экспериментально наблюдаемой зависимости, согласно которой в области pH 3-9,5 скорость постоянна и убывает при рН > 10. Таким образом, простейшая кинетическая модель объясняет некоторые эффекты, но должна быть усовершенствована для того, чтобы описать зависимости реакции от концентрации ионов водорода.
Кинетические модели, объясняющие зависимость реакции от pH. Чтобы объяснить зависимость максимальной скорости реакции от концентрации ионов водорода, необходимо постулировать наличие некоторых дополнительных стадий в обсуждаемых выше простейших механизмах реакций с участием гидрогеназ. "Насыщение" зависимости максимальной скорости реакции выделения водорода при высоких значениях pH может быть описано механизмом, включающим стадию образования промежуточного соединения фермента с протонами:
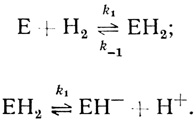 (2.42)
(2.42)При рассмотрении кинетики реакции в обратном направлении это соответствует механизму Михаэлиса-Ментен, который постулирует быстрое равновесное образование промежуточного фермент-субстратного комплекса (ионы водорода выступают в реакциях, катализируемых гидрогеназами, в качестве субстратов).
В работе [327] проведен анализ также группы механизмов, включающих параллельную реакцию с образованием нереакционноспособного комплекса.
Результаты кинетического анализа схем, включающих стадии (2.42), сведены в табл. 19. Механизмы представляют собой соответственно модификации механизмов ере, ерр и рее.
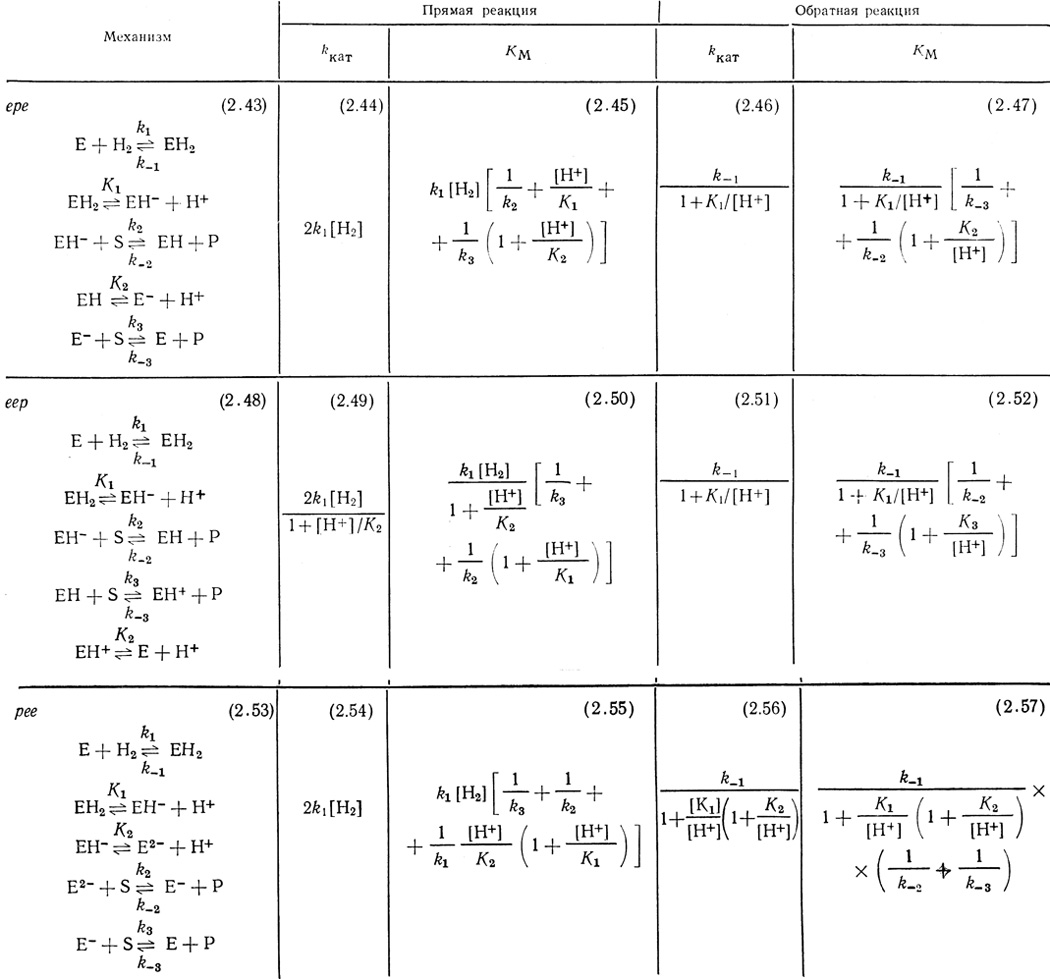
Таблица 19
В таблице 19 приведены выражения для параметров kкат и КМ, соответствующих начальным скоростям прямой и обратной реакций для изученных механизмов. Рассчитанные функции kкат могут быть сопоставлены с экспериментальными. Важно подчеркнуть, что зависимость kкат от pH для обратной реакции представляет собой функцию с "насыщением" и описывает экспериментально полученные результаты.
Сопоставление кинетических моделей с экспериментальными данными. Рассмотренные кинетические модели отражают наиболее существенные черты кинетических закономерностей катализа гидрогеназами.
1. Уравнения скорости для прямой и обратной реакций с участием гидрогеназы имеет "классический" для ферментативных реакций вид. Зависимости скорости реакции от концентраций органических субстратов даны в виде уравнения Михаэлиса. Важно отметить, что все исследованные механизмы не включают стадию образования "комплекса Михаэлиса", тем не менее уравнение скорости реакций имеет вид уравнения Михаэлиса-Ментен. В случае гидрогеназы уравнение Михаэлиса отражает тот факт, что при "высоких", "насыщающих" концентрациях донора (или акцептора) электрона лимитирующей становится стадия образования фермент-гидридного комплекса. При сравнении кинетических механизмов катализа гидрогеназами удобным параметром для анализа является величина kкат (или υмакс).
В то время как величины Км, найденные для различных механизмов, выражаются сложными функциями констант скорости и равновесия всех стадий процесса, величины kкат даны относительно простыми функциями. При этом надо иметь в виду, что на зависимость величины КМ от pH может накладывать отпечаток процесс ионизации субстрата. Нами было показано, что такие субстраты, как метилвиологен и бензилвиологен, в условиях опыта претерпевают некоторые превращения с рКа 6,5 [328]. Нейтральный красный и его восстановленная форма существуют в различных протонированных формах, и соотношения их концентраций зависят от pH среды. Поэтому величина kкат - наиболее прямой параметр в исследованных каталитических реакциях с участием гидрогеназы.
2. Важной общей особенностью обсуждаемых кинетических механизмов является тот факт, что величина kкат как прямой, так и обратной реакций определяется константами скорости взаимодействия фермента с водородом и константами равновесия реакций протонирования - депротонирования активного центра фермента. При этом параметр kкат не включает константы скорости переноса электрона на органический субстрат (или от него). Это количественно проявляется в том, что величина kкат (или υмакс) не зависят от природы используемого органического субстрата.
3. Кинетические модели достаточно хорошо описывают экспериментально найденные зависимости скорости реакций от концентраций ионов водорода.
4. Существенным достижением проведенного кинетического анализа является возможность классификации окислительно-восстановительных реакций по последовательности стадий переноса электрона и протона. Очевидно, механизмы реакций с двухэлектронным переносом на разных стадиях должны кинетически различаться. Для механизма катализа гидрогеназой реакции восстановления одноэлектронных акцепторов (или в обратной реакции - окисления одноэлектронных доноров) это проявляется наиболее ярко. В реакции с участием гидрогеназы перенос электронов и протонов из фермент-гидридного соединения может проходить в различных последовательностях: ере, рее или еер. При кинетическом анализе в принципе можно разграничить эти механизмы. Механизмы еер, рее и ере различаются по зависимости параметров kкат и КМ от pH (см. табл. 18 и 19). К сожалению, механизмы реакций с участием исследуемых субстратов осложнены pH-эффектами, связанными с ионизацией субстратов, что в свою очередь усложняет дискриминацию механизмов.
Константы скорости "элементарных" стадий и профиль свободной энергии катализа гидрогеназой. На основе проведенного кинетического анализа интересно сделать некоторые количественные оценки. Прежде всего из полученных экспериментальных данных можно найти константу скорости распада фермент-гидридного комплекса k-1 и константу равновесия его депротонирования К1. Во всех рассмотренных механизмах, объясняющих рН-зависимость (см. табл. 19), выражение для kкат (или υмакс) обратной реакции имеет один и тот же вид (см. уравнения (2.46), (2.51), (2.56)). На основе значения kкат (при [H+] >> K1) в реакции выделения водорода и исследования pH-зависимости этой константы величина k-1 равна 3,8 с-1 и К1 = 10-10 М.
Из уравнений, приведенных в табл. 19, следует, что kкат для реакции в прямом направлении определяется константой скорости k1 и, кроме того, константами равновесия протолитических равновесий промежуточных соединений. Поэтому численное значение константы k1 найденное из величины kкат (в прямом направлении), должно зависеть от выбора конкретного механизма. В настоящее время отсутствуют четкие критерии, которые позволили бы сделать этот выбор. Тем не менее для одного из механизмов нами сделаны количественные оценки для определения порядка величины и константы равновесия при образовании фермент-гидридного комплекса. Такого рода оценки чрезвычайно важны для понимания механизма катализа ферментом.
Из общих соображений механизм (2.43) (механизм ере с последовательным протонированием) представляется наиболее вероятным. Использование известной из литературы концентрации растворимого водорода [Н2] = 8⋅10-4 М уравнение (2.44) дает значение k1 = 600 М-1⋅с-1. Соответственно значение константы равновесия при образовании фермент-гидридного комплекса К = k-1/k1, полученное из этих данных, равно 6,3⋅10-3 М.
В соответствии с теорией переходного состояния полученные данные позволяют построить профиль изменений свободной энергии при протекании каталитического процесса. Профиль свободной энергии в механизме катализа гидрогеназой представлен на рис. 17. Минимумы свободной энергии по механизму реакции (2.43) соответствуют промежуточным соединениям ЕН2, ЕН-, ЕН и Е-. Из полученных кинетических данных можно получить информацию об интермедиатах ЕН2 и ЕН-. Именно стадии, приводящие к образованию этих интермедиатов, представляют главный интерес, поскольку связаны с активацией молекулярного водорода.
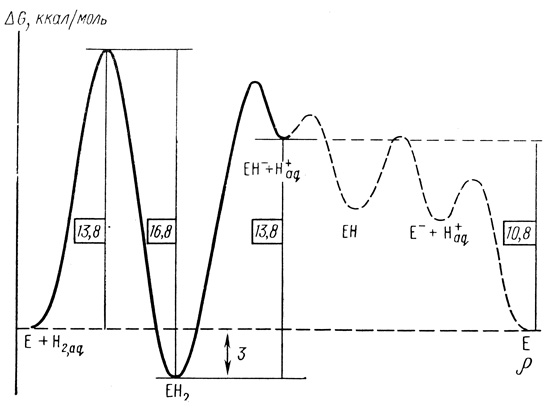
Рис. 17. Профиль свободной энергии в механизме катализа гидрогеназой
Из данных рис. 17 следует, что активация водорода гидрогеназой включает термодинамически невыгодное образование фермент-гидридного комплекса ЕН-, которое идет с затратами свободной энергии ∼10,7 ккал/моль. Для сравнения укажем, что теплота гомолитического расщепления молекулы водорода на атомы в водной среде равна 104,5 ккал/моль, свободная энергия гетеролитического расщепления с образованием гидрид-иона и протона равна 33 ккал/моль [335]. Таким образом, фермент выбирает путь реакции без образования термодинамически очень невыгодных промежуточных частиц, таких как атомы водорода. При этом активный центр существенно стабилизирует гидрид-ион (на 23 ккал/моль по сравнению с реакцией в воде).
Молекулярная модель механизма катализа гидрогеназой. Проведенное исследование кинетики действия гидрогеназы позволяет высказать некоторые предположения о молекулярных механизмах трансформации веществ под действием катализатора. Известно, что активный центр гидрогеназы из Th. roseopersicina включает четыре иона железа и четыре иона кислотолабильной серы [304] (предположительно 4Fe-4S-тип). Возможно, что ионы железа и серы объединены в комплекс кластерного типа, характерный для большинства железосерных негеминовых белков. Элементарные акты трансформации молекулы водорода и стадии переноса электрона должны осуществляться с участием этих комплексов.
Связи ионов железа с ионами серы в обсуждаемых структурах имеют, по-видимому, ионный или сильно полярный характер. Можно думать, что именно ионная связь Fe-S и есть тот полярный активный центр, который осуществляет гетеролитическое расщепление молекулы водорода. Можно представить себе следующий молекулярный механизм действия гидрогеназы:
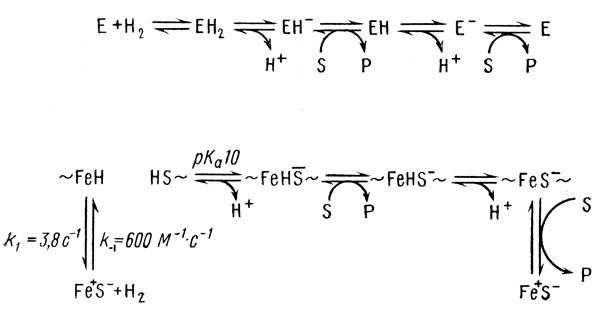 (2.58)
(2.58)Этот механизм основан на схеме (2.43). Ион железа в предлагаемом механизме играет роль акцептора гидрида, а ион серы сильного основания, акцептирующего протон. Последующие стадии представляют собой реакции акцептирования электрона, протона и электрона (ере-механизм).
|
ПОИСК:
|
При копировании материалов проекта обязательно ставить активную ссылку на страницу источник:
http://chemlib.ru/ 'Библиотека по химии'